Get trending papers in your email inbox once a day!
Get trending papers in your email inbox!
Subscribe
Enhancing Score-Based Sampling Methods with Ensembles
We introduce ensembles within score-based sampling methods to develop gradient-free approximate sampling techniques that leverage the collective dynamics of particle ensembles to compute approximate reverse diffusion drifts. We introduce the underlying methodology, emphasizing its relationship with generative diffusion models and the previously introduced F\"ollmer sampler. We demonstrate the efficacy of ensemble strategies through various examples, ranging from low- to medium-dimensionality sampling problems, including multi-modal and highly non-Gaussian probability distributions, and provide comparisons to traditional methods like NUTS. Our findings highlight the potential of ensemble strategies for modeling complex probability distributions in situations where gradients are unavailable. Finally, we showcase its application in the context of Bayesian inversion problems within the geophysical sciences.
AutoDES: AutoML Pipeline Generation of Classification with Dynamic Ensemble Strategy Selection
Automating machine learning has achieved remarkable technological developments in recent years, and building an automated machine learning pipeline is now an essential task. The model ensemble is the technique of combining multiple models to get a better and more robust model. However, existing automated machine learning tends to be simplistic in handling the model ensemble, where the ensemble strategy is fixed, such as stacked generalization. There have been many techniques on different ensemble methods, especially ensemble selection, and the fixed ensemble strategy limits the upper limit of the model's performance. In this article, we present a novel framework for automated machine learning. Our framework incorporates advances in dynamic ensemble selection, and to our best knowledge, our approach is the first in the field of AutoML to search and optimize ensemble strategies. In the comparison experiments, our method outperforms the state-of-the-art automated machine learning frameworks with the same CPU time in 42 classification datasets from the OpenML platform. Ablation experiments on our framework validate the effectiveness of our proposed method.
Characterizing gaussian mixture of motion modes for skid-steer state estimation
Skid-steered wheel mobile robots (SSWMRs) are characterized by the unique domination of the tire-terrain skidding for the robot to move. The lack of reliable friction models cascade into unreliable motion models, especially the reduced ordered variants used for state estimation and robot control. Ensemble modeling is an emerging research direction where the overall motion model is broken down into a family of local models to distribute the performance and resource requirement and provide a fast real-time prediction. To this end, a gaussian mixture model based modeling identification of model clusters is adopted and implemented within an interactive multiple model (IMM) based state estimation. The framework is adopted and implemented for angular velocity as the estimated state for a mid scaled skid-steered wheel mobile robot platform.
Input-gradient space particle inference for neural network ensembles
Deep Ensembles (DEs) demonstrate improved accuracy, calibration and robustness to perturbations over single neural networks partly due to their functional diversity. Particle-based variational inference (ParVI) methods enhance diversity by formalizing a repulsion term based on a network similarity kernel. However, weight-space repulsion is inefficient due to over-parameterization, while direct function-space repulsion has been found to produce little improvement over DEs. To sidestep these difficulties, we propose First-order Repulsive Deep Ensemble (FoRDE), an ensemble learning method based on ParVI, which performs repulsion in the space of first-order input gradients. As input gradients uniquely characterize a function up to translation and are much smaller in dimension than the weights, this method guarantees that ensemble members are functionally different. Intuitively, diversifying the input gradients encourages each network to learn different features, which is expected to improve the robustness of an ensemble. Experiments on image classification datasets and transfer learning tasks show that FoRDE significantly outperforms the gold-standard DEs and other ensemble methods in accuracy and calibration under covariate shift due to input perturbations.
Differentiable Model Selection for Ensemble Learning
Model selection is a strategy aimed at creating accurate and robust models. A key challenge in designing these algorithms is identifying the optimal model for classifying any particular input sample. This paper addresses this challenge and proposes a novel framework for differentiable model selection integrating machine learning and combinatorial optimization. The framework is tailored for ensemble learning, a strategy that combines the outputs of individually pre-trained models, and learns to select appropriate ensemble members for a particular input sample by transforming the ensemble learning task into a differentiable selection program trained end-to-end within the ensemble learning model. Tested on various tasks, the proposed framework demonstrates its versatility and effectiveness, outperforming conventional and advanced consensus rules across a variety of settings and learning tasks.

Window-Based Early-Exit Cascades for Uncertainty Estimation: When Deep Ensembles are More Efficient than Single Models
Deep Ensembles are a simple, reliable, and effective method of improving both the predictive performance and uncertainty estimates of deep learning approaches. However, they are widely criticised as being computationally expensive, due to the need to deploy multiple independent models. Recent work has challenged this view, showing that for predictive accuracy, ensembles can be more computationally efficient (at inference) than scaling single models within an architecture family. This is achieved by cascading ensemble members via an early-exit approach. In this work, we investigate extending these efficiency gains to tasks related to uncertainty estimation. As many such tasks, e.g. selective classification, are binary classification, our key novel insight is to only pass samples within a window close to the binary decision boundary to later cascade stages. Experiments on ImageNet-scale data across a number of network architectures and uncertainty tasks show that the proposed window-based early-exit approach is able to achieve a superior uncertainty-computation trade-off compared to scaling single models. For example, a cascaded EfficientNet-B2 ensemble is able to achieve similar coverage at 5% risk as a single EfficientNet-B4 with <30% the number of MACs. We also find that cascades/ensembles give more reliable improvements on OOD data vs scaling models up. Code for this work is available at: https://github.com/Guoxoug/window-early-exit.

Enhancing Neural Subset Selection: Integrating Background Information into Set Representations
Learning neural subset selection tasks, such as compound selection in AI-aided drug discovery, have become increasingly pivotal across diverse applications. The existing methodologies in the field primarily concentrate on constructing models that capture the relationship between utility function values and subsets within their respective supersets. However, these approaches tend to overlook the valuable information contained within the superset when utilizing neural networks to model set functions. In this work, we address this oversight by adopting a probabilistic perspective. Our theoretical findings demonstrate that when the target value is conditioned on both the input set and subset, it is essential to incorporate an invariant sufficient statistic of the superset into the subset of interest for effective learning. This ensures that the output value remains invariant to permutations of the subset and its corresponding superset, enabling identification of the specific superset from which the subset originated. Motivated by these insights, we propose a simple yet effective information aggregation module designed to merge the representations of subsets and supersets from a permutation invariance perspective. Comprehensive empirical evaluations across diverse tasks and datasets validate the enhanced efficacy of our approach over conventional methods, underscoring the practicality and potency of our proposed strategies in real-world contexts.

Spurious Feature Diversification Improves Out-of-distribution Generalization
Generalization to out-of-distribution (OOD) data is a critical challenge in machine learning. Ensemble-based methods, like weight space ensembles that interpolate model parameters, have been shown to achieve superior OOD performance. However, the underlying mechanism for their effectiveness remains unclear. In this study, we closely examine WiSE-FT, a popular weight space ensemble method that interpolates between a pre-trained and a fine-tuned model. We observe an unexpected phenomenon, in which WiSE-FT successfully corrects many cases where each individual model makes incorrect predictions, which contributes significantly to its OOD effectiveness. To gain further insights, we conduct theoretical analysis in a multi-class setting with a large number of spurious features. Our analysis predicts the above phenomenon and it further shows that ensemble-based models reduce prediction errors in the OOD settings by utilizing a more diverse set of spurious features. Contrary to the conventional wisdom that focuses on learning invariant features for better OOD performance, our findings suggest that incorporating a large number of diverse spurious features weakens their individual contributions, leading to improved overall OOD generalization performance. Empirically we demonstrate the effectiveness of utilizing diverse spurious features on a MultiColorMNIST dataset, and our experimental results are consistent with the theoretical analysis. Building upon the new theoretical insights into the efficacy of ensemble methods, we further identify an issue of WiSE-FT caused by the overconfidence of fine-tuned models in OOD situations. This overconfidence magnifies the fine-tuned model's incorrect prediction, leading to deteriorated OOD ensemble performance. To remedy this problem, we propose a novel method called BAlaNced averaGing (BANG), which significantly enhances the OOD performance of WiSE-FT.

Matrix approach to generalized ensemble theory
We provide a concise framework for generalized ensemble theory through a matrix-based approach. By introducing an observation matrix, any discrete probability distribution, including those for non-equilibrium steady states, can be expressed as a generalized Boltzmann distribution, with observables and conjugate variables as the basis and coordinates in a linear space. In this framework, we identify the minimal sufficient statistics required for inferring the Boltzmann distribution. Furthermore, we show that the Hadamard and Vandermonde matrices are suitable observation matrices for spin systems and random walks. In master equation systems, the probability flux observation matrix facilitates the identification of detailed balance violations. Our findings provide a new approach to developing generalized ensemble theory for non-equilibrium steady-state systems.
AutoDEUQ: Automated Deep Ensemble with Uncertainty Quantification
Deep neural networks are powerful predictors for a variety of tasks. However, they do not capture uncertainty directly. Using neural network ensembles to quantify uncertainty is competitive with approaches based on Bayesian neural networks while benefiting from better computational scalability. However, building ensembles of neural networks is a challenging task because, in addition to choosing the right neural architecture or hyperparameters for each member of the ensemble, there is an added cost of training each model. We propose AutoDEUQ, an automated approach for generating an ensemble of deep neural networks. Our approach leverages joint neural architecture and hyperparameter search to generate ensembles. We use the law of total variance to decompose the predictive variance of deep ensembles into aleatoric (data) and epistemic (model) uncertainties. We show that AutoDEUQ outperforms probabilistic backpropagation, Monte Carlo dropout, deep ensemble, distribution-free ensembles, and hyper ensemble methods on a number of regression benchmarks.


Selective Ensembles for Consistent Predictions
Recent work has shown that models trained to the same objective, and which achieve similar measures of accuracy on consistent test data, may nonetheless behave very differently on individual predictions. This inconsistency is undesirable in high-stakes contexts, such as medical diagnosis and finance. We show that this inconsistent behavior extends beyond predictions to feature attributions, which may likewise have negative implications for the intelligibility of a model, and one's ability to find recourse for subjects. We then introduce selective ensembles to mitigate such inconsistencies by applying hypothesis testing to the predictions of a set of models trained using randomly-selected starting conditions; importantly, selective ensembles can abstain in cases where a consistent outcome cannot be achieved up to a specified confidence level. We prove that that prediction disagreement between selective ensembles is bounded, and empirically demonstrate that selective ensembles achieve consistent predictions and feature attributions while maintaining low abstention rates. On several benchmark datasets, selective ensembles reach zero inconsistently predicted points, with abstention rates as low 1.5%.
Distributional Reinforcement Learning with Ensembles
It is well known that ensemble methods often provide enhanced performance in reinforcement learning. In this paper, we explore this concept further by using group-aided training within the distributional reinforcement learning paradigm. Specifically, we propose an extension to categorical reinforcement learning, where distributional learning targets are implicitly based on the total information gathered by an ensemble. We empirically show that this may lead to much more robust initial learning, a stronger individual performance level, and good efficiency on a per-sample basis.

Pathologies of Predictive Diversity in Deep Ensembles
Classic results establish that encouraging predictive diversity improves performance in ensembles of low-capacity models, e.g. through bagging or boosting. Here we demonstrate that these intuitions do not apply to high-capacity neural network ensembles (deep ensembles), and in fact the opposite is often true. In a large scale study of nearly 600 neural network classification ensembles, we examine a variety of interventions that trade off component model performance for predictive diversity. While such interventions can improve the performance of small neural network ensembles (in line with standard intuitions), they harm the performance of the large neural network ensembles most often used in practice. Surprisingly, we also find that discouraging predictive diversity is often benign in large-network ensembles, fully inverting standard intuitions. Even when diversity-promoting interventions do not sacrifice component model performance (e.g. using heterogeneous architectures and training paradigms), we observe an opportunity cost associated with pursuing increased predictive diversity. Examining over 1000 ensembles, we observe that the performance benefits of diverse architectures/training procedures are easily dwarfed by the benefits of simply using higher-capacity models, despite the fact that such higher capacity models often yield significantly less predictive diversity. Overall, our findings demonstrate that standard intuitions around predictive diversity, originally developed for low-capacity ensembles, do not directly apply to modern high-capacity deep ensembles. This work clarifies fundamental challenges to the goal of improving deep ensembles by making them more diverse, while suggesting an alternative path: simply forming ensembles from ever more powerful (and less diverse) component models.

Exact Learning of Permutations for Nonzero Binary Inputs with Logarithmic Training Size and Quadratic Ensemble Complexity
The ability of an architecture to realize permutations is quite fundamental. For example, Large Language Models need to be able to correctly copy (and perhaps rearrange) parts of the input prompt into the output. Classical universal approximation theorems guarantee the existence of parameter configurations that solve this task but offer no insights into whether gradient-based algorithms can find them. In this paper, we address this gap by focusing on two-layer fully connected feed-forward neural networks and the task of learning permutations on nonzero binary inputs. We show that in the infinite width Neural Tangent Kernel (NTK) regime, an ensemble of such networks independently trained with gradient descent on only the k standard basis vectors out of 2^k - 1 possible inputs successfully learns any fixed permutation of length k with arbitrarily high probability. By analyzing the exact training dynamics, we prove that the network's output converges to a Gaussian process whose mean captures the ground truth permutation via sign-based features. We then demonstrate how averaging these runs (an "ensemble" method) and applying a simple rounding step yields an arbitrarily accurate prediction on any possible input unseen during training. Notably, the number of models needed to achieve exact learning with high probability (which we refer to as ensemble complexity) exhibits a linearithmic dependence on the input size k for a single test input and a quadratic dependence when considering all test inputs simultaneously.

Mechanistic Mode Connectivity
We study neural network loss landscapes through the lens of mode connectivity, the observation that minimizers of neural networks retrieved via training on a dataset are connected via simple paths of low loss. Specifically, we ask the following question: are minimizers that rely on different mechanisms for making their predictions connected via simple paths of low loss? We provide a definition of mechanistic similarity as shared invariances to input transformations and demonstrate that lack of linear connectivity between two models implies they use dissimilar mechanisms for making their predictions. Relevant to practice, this result helps us demonstrate that naive fine-tuning on a downstream dataset can fail to alter a model's mechanisms, e.g., fine-tuning can fail to eliminate a model's reliance on spurious attributes. Our analysis also motivates a method for targeted alteration of a model's mechanisms, named connectivity-based fine-tuning (CBFT), which we analyze using several synthetic datasets for the task of reducing a model's reliance on spurious attributes.


DivBO: Diversity-aware CASH for Ensemble Learning
The Combined Algorithm Selection and Hyperparameters optimization (CASH) problem is one of the fundamental problems in Automated Machine Learning (AutoML). Motivated by the success of ensemble learning, recent AutoML systems build post-hoc ensembles to output the final predictions instead of using the best single learner. However, while most CASH methods focus on searching for a single learner with the best performance, they neglect the diversity among base learners (i.e., they may suggest similar configurations to previously evaluated ones), which is also a crucial consideration when building an ensemble. To tackle this issue and further enhance the ensemble performance, we propose DivBO, a diversity-aware framework to inject explicit search of diversity into the CASH problems. In the framework, we propose to use a diversity surrogate to predict the pair-wise diversity of two unseen configurations. Furthermore, we introduce a temporary pool and a weighted acquisition function to guide the search of both performance and diversity based on Bayesian optimization. Empirical results on 15 public datasets show that DivBO achieves the best average ranks (1.82 and 1.73) on both validation and test errors among 10 compared methods, including post-hoc designs in recent AutoML systems and state-of-the-art baselines for ensemble learning on CASH problems.
Which Invariance Should We Transfer? A Causal Minimax Learning Approach
A major barrier to deploying current machine learning models lies in their non-reliability to dataset shifts. To resolve this problem, most existing studies attempted to transfer stable information to unseen environments. Particularly, independent causal mechanisms-based methods proposed to remove mutable causal mechanisms via the do-operator. Compared to previous methods, the obtained stable predictors are more effective in identifying stable information. However, a key question remains: which subset of this whole stable information should the model transfer, in order to achieve optimal generalization ability? To answer this question, we present a comprehensive minimax analysis from a causal perspective. Specifically, we first provide a graphical condition for the whole stable set to be optimal. When this condition fails, we surprisingly find with an example that this whole stable set, although can fully exploit stable information, is not the optimal one to transfer. To identify the optimal subset under this case, we propose to estimate the worst-case risk with a novel optimization scheme over the intervention functions on mutable causal mechanisms. We then propose an efficient algorithm to search for the subset with minimal worst-case risk, based on a newly defined equivalence relation between stable subsets. Compared to the exponential cost of exhaustively searching over all subsets, our searching strategy enjoys a polynomial complexity. The effectiveness and efficiency of our methods are demonstrated on synthetic data and the diagnosis of Alzheimer's disease.
Deep Ensembles Work, But Are They Necessary?
Ensembling neural networks is an effective way to increase accuracy, and can often match the performance of individual larger models. This observation poses a natural question: given the choice between a deep ensemble and a single neural network with similar accuracy, is one preferable over the other? Recent work suggests that deep ensembles may offer distinct benefits beyond predictive power: namely, uncertainty quantification and robustness to dataset shift. In this work, we demonstrate limitations to these purported benefits, and show that a single (but larger) neural network can replicate these qualities. First, we show that ensemble diversity, by any metric, does not meaningfully contribute to an ensemble's uncertainty quantification on out-of-distribution (OOD) data, but is instead highly correlated with the relative improvement of a single larger model. Second, we show that the OOD performance afforded by ensembles is strongly determined by their in-distribution (InD) performance, and -- in this sense -- is not indicative of any "effective robustness". While deep ensembles are a practical way to achieve improvements to predictive power, uncertainty quantification, and robustness, our results show that these improvements can be replicated by a (larger) single model.
Q(D)O-ES: Population-based Quality (Diversity) Optimisation for Post Hoc Ensemble Selection in AutoML
Automated machine learning (AutoML) systems commonly ensemble models post hoc to improve predictive performance, typically via greedy ensemble selection (GES). However, we believe that GES may not always be optimal, as it performs a simple deterministic greedy search. In this work, we introduce two novel population-based ensemble selection methods, QO-ES and QDO-ES, and compare them to GES. While QO-ES optimises solely for predictive performance, QDO-ES also considers the diversity of ensembles within the population, maintaining a diverse set of well-performing ensembles during optimisation based on ideas of quality diversity optimisation. The methods are evaluated using 71 classification datasets from the AutoML benchmark, demonstrating that QO-ES and QDO-ES often outrank GES, albeit only statistically significant on validation data. Our results further suggest that diversity can be beneficial for post hoc ensembling but also increases the risk of overfitting.
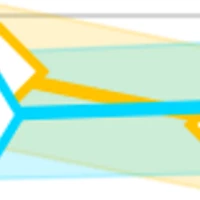
JAMUN: Bridging Smoothed Molecular Dynamics and Score-Based Learning for Conformational Ensembles
Conformational ensembles of protein structures are immensely important both for understanding protein function and drug discovery in novel modalities such as cryptic pockets. Current techniques for sampling ensembles such as molecular dynamics (MD) are computationally inefficient, while many recent machine learning methods do not transfer to systems outside their training data. We propose JAMUN which performs MD in a smoothed, noised space of all-atom 3D conformations of molecules by utilizing the framework of walk-jump sampling. JAMUN enables ensemble generation for small peptides at rates of an order of magnitude faster than traditional molecular dynamics. The physical priors in JAMUN enables transferability to systems outside of its training data, even to peptides that are longer than those originally trained on. Our model, code and weights are available at https://github.com/prescient-design/jamun.
Estimating Causal Effects using a Multi-task Deep Ensemble
A number of methods have been proposed for causal effect estimation, yet few have demonstrated efficacy in handling data with complex structures, such as images. To fill this gap, we propose Causal Multi-task Deep Ensemble (CMDE), a novel framework that learns both shared and group-specific information from the study population. We provide proofs demonstrating equivalency of CDME to a multi-task Gaussian process (GP) with a coregionalization kernel a priori. Compared to multi-task GP, CMDE efficiently handles high-dimensional and multi-modal covariates and provides pointwise uncertainty estimates of causal effects. We evaluate our method across various types of datasets and tasks and find that CMDE outperforms state-of-the-art methods on a majority of these tasks.


P2DFlow: A Protein Ensemble Generative Model with SE(3) Flow Matching
Biological processes, functions, and properties are intricately linked to the ensemble of protein conformations, rather than being solely determined by a single stable conformation. In this study, we have developed P2DFlow, a generative model based on SE(3) flow matching, to predict the structural ensembles of proteins. We specifically designed a valuable prior for the flow process and enhanced the model's ability to distinguish each intermediate state by incorporating an additional dimension to describe the ensemble data, which can reflect the physical laws governing the distribution of ensembles, so that the prior knowledge can effectively guide the generation process. When trained and evaluated on the MD datasets of ATLAS, P2DFlow outperforms other baseline models on extensive experiments, successfully capturing the observable dynamic fluctuations as evidenced in crystal structure and MD simulations. As a potential proxy agent for protein molecular simulation, the high-quality ensembles generated by P2DFlow could significantly aid in understanding protein functions across various scenarios. Code is available at https://github.com/BLEACH366/P2DFlow

Universal Neurons in GPT2 Language Models
A basic question within the emerging field of mechanistic interpretability is the degree to which neural networks learn the same underlying mechanisms. In other words, are neural mechanisms universal across different models? In this work, we study the universality of individual neurons across GPT2 models trained from different initial random seeds, motivated by the hypothesis that universal neurons are likely to be interpretable. In particular, we compute pairwise correlations of neuron activations over 100 million tokens for every neuron pair across five different seeds and find that 1-5\% of neurons are universal, that is, pairs of neurons which consistently activate on the same inputs. We then study these universal neurons in detail, finding that they usually have clear interpretations and taxonomize them into a small number of neuron families. We conclude by studying patterns in neuron weights to establish several universal functional roles of neurons in simple circuits: deactivating attention heads, changing the entropy of the next token distribution, and predicting the next token to (not) be within a particular set.

Scalable Equilibrium Sampling with Sequential Boltzmann Generators
Scalable sampling of molecular states in thermodynamic equilibrium is a long-standing challenge in statistical physics. Boltzmann generators tackle this problem by pairing normalizing flows with importance sampling to obtain uncorrelated samples under the target distribution. In this paper, we extend the Boltzmann generator framework with two key contributions, denoting our framework Sequential Boltzmann Generators (SBG). The first is a highly efficient Transformer-based normalizing flow operating directly on all-atom Cartesian coordinates. In contrast to the equivariant continuous flows of prior methods, we leverage exactly invertible non-equivariant architectures which are highly efficient during both sample generation and likelihood evaluation. This efficiency unlocks more sophisticated inference strategies beyond standard importance sampling. In particular, we perform inference-time scaling of flow samples using a continuous-time variant of sequential Monte Carlo, in which flow samples are transported towards the target distribution with annealed Langevin dynamics. SBG achieves state-of-the-art performance w.r.t. all metrics on peptide systems, demonstrating the first equilibrium sampling in Cartesian coordinates of tri-, tetra- and hexa-peptides that were thus far intractable for prior Boltzmann generators.
Revisiting Ensemble Methods for Stock Trading and Crypto Trading Tasks at ACM ICAIF FinRL Contest 2023-2024
Reinforcement learning has demonstrated great potential for performing financial tasks. However, it faces two major challenges: policy instability and sampling bottlenecks. In this paper, we revisit ensemble methods with massively parallel simulations on graphics processing units (GPUs), significantly enhancing the computational efficiency and robustness of trained models in volatile financial markets. Our approach leverages the parallel processing capability of GPUs to significantly improve the sampling speed for training ensemble models. The ensemble models combine the strengths of component agents to improve the robustness of financial decision-making strategies. We conduct experiments in both stock and cryptocurrency trading tasks to evaluate the effectiveness of our approach. Massively parallel simulation on a single GPU improves the sampling speed by up to 1,746times using 2,048 parallel environments compared to a single environment. The ensemble models have high cumulative returns and outperform some individual agents, reducing maximum drawdown by up to 4.17% and improving the Sharpe ratio by up to 0.21. This paper describes trading tasks at ACM ICAIF FinRL Contests in 2023 and 2024.
Stochastic Normalizing Flows
The sampling of probability distributions specified up to a normalization constant is an important problem in both machine learning and statistical mechanics. While classical stochastic sampling methods such as Markov Chain Monte Carlo (MCMC) or Langevin Dynamics (LD) can suffer from slow mixing times there is a growing interest in using normalizing flows in order to learn the transformation of a simple prior distribution to the given target distribution. Here we propose a generalized and combined approach to sample target densities: Stochastic Normalizing Flows (SNF) -- an arbitrary sequence of deterministic invertible functions and stochastic sampling blocks. We show that stochasticity overcomes expressivity limitations of normalizing flows resulting from the invertibility constraint, whereas trainable transformations between sampling steps improve efficiency of pure MCMC/LD along the flow. By invoking ideas from non-equilibrium statistical mechanics we derive an efficient training procedure by which both the sampler's and the flow's parameters can be optimized end-to-end, and by which we can compute exact importance weights without having to marginalize out the randomness of the stochastic blocks. We illustrate the representational power, sampling efficiency and asymptotic correctness of SNFs on several benchmarks including applications to sampling molecular systems in equilibrium.
Why do Random Forests Work? Understanding Tree Ensembles as Self-Regularizing Adaptive Smoothers
Despite their remarkable effectiveness and broad application, the drivers of success underlying ensembles of trees are still not fully understood. In this paper, we highlight how interpreting tree ensembles as adaptive and self-regularizing smoothers can provide new intuition and deeper insight to this topic. We use this perspective to show that, when studied as smoothers, randomized tree ensembles not only make predictions that are quantifiably more smooth than the predictions of the individual trees they consist of, but also further regulate their smoothness at test-time based on the dissimilarity between testing and training inputs. First, we use this insight to revisit, refine and reconcile two recent explanations of forest success by providing a new way of quantifying the conjectured behaviors of tree ensembles objectively by measuring the effective degree of smoothing they imply. Then, we move beyond existing explanations for the mechanisms by which tree ensembles improve upon individual trees and challenge the popular wisdom that the superior performance of forests should be understood as a consequence of variance reduction alone. We argue that the current high-level dichotomy into bias- and variance-reduction prevalent in statistics is insufficient to understand tree ensembles -- because the prevailing definition of bias does not capture differences in the expressivity of the hypothesis classes formed by trees and forests. Instead, we show that forests can improve upon trees by three distinct mechanisms that are usually implicitly entangled. In particular, we demonstrate that the smoothing effect of ensembling can reduce variance in predictions due to noise in outcome generation, reduce variability in the quality of the learned function given fixed input data and reduce potential bias in learnable functions by enriching the available hypothesis space.
GeoGraph: Geometric and Graph-based Ensemble Descriptors for Intrinsically Disordered Proteins
While deep learning has revolutionized the prediction of rigid protein structures, modelling the conformational ensembles of Intrinsically Disordered Proteins (IDPs) remains a key frontier. Current AI paradigms present a trade-off: Protein Language Models (PLMs) capture evolutionary statistics but lack explicit physical grounding, while generative models trained to model full ensembles are computationally expensive. In this work we critically assess these limits and propose a path forward. We introduce GeoGraph, a simulation-informed surrogate trained to predict ensemble-averaged statistics of residue-residue contact-map topology directly from sequence. By featurizing coarse-grained molecular dynamics simulations into residue- and sequence-level graph descriptors, we create a robust and information-rich learning target. Our evaluation demonstrates that this approach yields representations that are more predictive of key biophysical properties than existing methods.
FEAT: Free energy Estimators with Adaptive Transport
We present Free energy Estimators with Adaptive Transport (FEAT), a novel framework for free energy estimation -- a critical challenge across scientific domains. FEAT leverages learned transports implemented via stochastic interpolants and provides consistent, minimum-variance estimators based on escorted Jarzynski equality and controlled Crooks theorem, alongside variational upper and lower bounds on free energy differences. Unifying equilibrium and non-equilibrium methods under a single theoretical framework, FEAT establishes a principled foundation for neural free energy calculations. Experimental validation on toy examples, molecular simulations, and quantum field theory demonstrates improvements over existing learning-based methods.
Attention-based Ensemble for Deep Metric Learning
Deep metric learning aims to learn an embedding function, modeled as deep neural network. This embedding function usually puts semantically similar images close while dissimilar images far from each other in the learned embedding space. Recently, ensemble has been applied to deep metric learning to yield state-of-the-art results. As one important aspect of ensemble, the learners should be diverse in their feature embeddings. To this end, we propose an attention-based ensemble, which uses multiple attention masks, so that each learner can attend to different parts of the object. We also propose a divergence loss, which encourages diversity among the learners. The proposed method is applied to the standard benchmarks of deep metric learning and experimental results show that it outperforms the state-of-the-art methods by a significant margin on image retrieval tasks.
Rethinking LLM Evaluation: Can We Evaluate LLMs with 200x Less Data?
As the demand for comprehensive evaluations of diverse model capabilities steadily increases, benchmark suites have correspondingly grown significantly in scale. Despite notable advances in redundancy reduction and subset-level performance prediction, a systematic framework that effectively integrates these methods to ensure both prediction accuracy and ranking consistency is still largely elusive. In this paper, we first perform a sample-level analysis of benchmark redundancy and identify several highly similar samples that can be eliminated. Besides, we frame benchmark compression as an optimization problem with the aim of score reconstruction. Building on these, we then propose EssenceBench, a coarse-to-fine framework utilizing an iterative Genetic Algorithm (GA), which takes the advantages of fitness-based subset search and attribution-based sample search. Compared to previous methods, our approach yields superior compression results with lower reconstruction error and markedly higher efficiency. In particular, on the HellaSwag benchmark (10K samples), our method preserves the ranking of all models shifting within 5% using 25x fewer samples, and achieves 95% ranking preservation shifting within 5% using only 200x fewer samples.

Generative Modeling of Molecular Dynamics Trajectories
Molecular dynamics (MD) is a powerful technique for studying microscopic phenomena, but its computational cost has driven significant interest in the development of deep learning-based surrogate models. We introduce generative modeling of molecular trajectories as a paradigm for learning flexible multi-task surrogate models of MD from data. By conditioning on appropriately chosen frames of the trajectory, we show such generative models can be adapted to diverse tasks such as forward simulation, transition path sampling, and trajectory upsampling. By alternatively conditioning on part of the molecular system and inpainting the rest, we also demonstrate the first steps towards dynamics-conditioned molecular design. We validate the full set of these capabilities on tetrapeptide simulations and show that our model can produce reasonable ensembles of protein monomers. Altogether, our work illustrates how generative modeling can unlock value from MD data towards diverse downstream tasks that are not straightforward to address with existing methods or even MD itself. Code is available at https://github.com/bjing2016/mdgen.
Deep Sets
We study the problem of designing models for machine learning tasks defined on sets. In contrast to traditional approach of operating on fixed dimensional vectors, we consider objective functions defined on sets that are invariant to permutations. Such problems are widespread, ranging from estimation of population statistics poczos13aistats, to anomaly detection in piezometer data of embankment dams Jung15Exploration, to cosmology Ntampaka16Dynamical,Ravanbakhsh16ICML1. Our main theorem characterizes the permutation invariant functions and provides a family of functions to which any permutation invariant objective function must belong. This family of functions has a special structure which enables us to design a deep network architecture that can operate on sets and which can be deployed on a variety of scenarios including both unsupervised and supervised learning tasks. We also derive the necessary and sufficient conditions for permutation equivariance in deep models. We demonstrate the applicability of our method on population statistic estimation, point cloud classification, set expansion, and outlier detection.
Action Matching: Learning Stochastic Dynamics from Samples
Learning the continuous dynamics of a system from snapshots of its temporal marginals is a problem which appears throughout natural sciences and machine learning, including in quantum systems, single-cell biological data, and generative modeling. In these settings, we assume access to cross-sectional samples that are uncorrelated over time, rather than full trajectories of samples. In order to better understand the systems under observation, we would like to learn a model of the underlying process that allows us to propagate samples in time and thereby simulate entire individual trajectories. In this work, we propose Action Matching, a method for learning a rich family of dynamics using only independent samples from its time evolution. We derive a tractable training objective, which does not rely on explicit assumptions about the underlying dynamics and does not require back-propagation through differential equations or optimal transport solvers. Inspired by connections with optimal transport, we derive extensions of Action Matching to learn stochastic differential equations and dynamics involving creation and destruction of probability mass. Finally, we showcase applications of Action Matching by achieving competitive performance in a diverse set of experiments from biology, physics, and generative modeling.

Scalable Bayesian Uncertainty Quantification for Neural Network Potentials: Promise and Pitfalls
Neural network (NN) potentials promise highly accurate molecular dynamics (MD) simulations within the computational complexity of classical MD force fields. However, when applied outside their training domain, NN potential predictions can be inaccurate, increasing the need for Uncertainty Quantification (UQ). Bayesian modeling provides the mathematical framework for UQ, but classical Bayesian methods based on Markov chain Monte Carlo (MCMC) are computationally intractable for NN potentials. By training graph NN potentials for coarse-grained systems of liquid water and alanine dipeptide, we demonstrate here that scalable Bayesian UQ via stochastic gradient MCMC (SG-MCMC) yields reliable uncertainty estimates for MD observables. We show that cold posteriors can reduce the required training data size and that for reliable UQ, multiple Markov chains are needed. Additionally, we find that SG-MCMC and the Deep Ensemble method achieve comparable results, despite shorter training and less hyperparameter tuning of the latter. We show that both methods can capture aleatoric and epistemic uncertainty reliably, but not systematic uncertainty, which needs to be minimized by adequate modeling to obtain accurate credible intervals for MD observables. Our results represent a step towards accurate UQ that is of vital importance for trustworthy NN potential-based MD simulations required for decision-making in practice.
MOOSE-Chem2: Exploring LLM Limits in Fine-Grained Scientific Hypothesis Discovery via Hierarchical Search
Large language models (LLMs) have shown promise in automating scientific hypothesis generation, yet existing approaches primarily yield coarse-grained hypotheses lacking critical methodological and experimental details. We introduce and formally define the novel task of fine-grained scientific hypothesis discovery, which entails generating detailed, experimentally actionable hypotheses from coarse initial research directions. We frame this as a combinatorial optimization problem and investigate the upper limits of LLMs' capacity to solve it when maximally leveraged. Specifically, we explore four foundational questions: (1) how to best harness an LLM's internal heuristics to formulate the fine-grained hypothesis it itself would judge as the most promising among all the possible hypotheses it might generate, based on its own internal scoring-thus defining a latent reward landscape over the hypothesis space; (2) whether such LLM-judged better hypotheses exhibit stronger alignment with ground-truth hypotheses; (3) whether shaping the reward landscape using an ensemble of diverse LLMs of similar capacity yields better outcomes than defining it with repeated instances of the strongest LLM among them; and (4) whether an ensemble of identical LLMs provides a more reliable reward landscape than a single LLM. To address these questions, we propose a hierarchical search method that incrementally proposes and integrates details into the hypothesis, progressing from general concepts to specific experimental configurations. We show that this hierarchical process smooths the reward landscape and enables more effective optimization. Empirical evaluations on a new benchmark of expert-annotated fine-grained hypotheses from recent chemistry literature show that our method consistently outperforms strong baselines.




HDEE: Heterogeneous Domain Expert Ensemble
Training dense LLMs requires enormous amounts of data and centralized compute, which introduces fundamental bottlenecks and ever-growing costs for large models. Several studies aim to reduce this dependency on centralization by reducing the communication overhead of training dense models. Taking this idea of reducing communication overhead to a natural extreme, by training embarrassingly parallelizable ensembles of small independent experts, has been shown to outperform large dense models trained in traditional centralized settings. However, existing studies do not take into account underlying differences amongst data domains and treat them as monolithic, regardless of their underlying complexity, size, or distribution. In this paper, we explore the effects of introducing heterogeneity to these ensembles of domain expert models. Specifically, by allowing models within the ensemble to vary in size--as well as the number of training steps taken depending on the training data's domain--we study the effect heterogeneity has on these ensembles when evaluated against domains included in, and excluded from, the training set. We use the same compute budget to train heterogeneous ensembles and homogeneous baselines for comparison. We show that the heterogeneous ensembles achieve the lowest perplexity scores in 20 out of the 21 data domains used in the evaluation. Our code is available at https://github.com/gensyn-ai/hdee.



Chemically Transferable Generative Backmapping of Coarse-Grained Proteins
Coarse-graining (CG) accelerates molecular simulations of protein dynamics by simulating sets of atoms as singular beads. Backmapping is the opposite operation of bringing lost atomistic details back from the CG representation. While machine learning (ML) has produced accurate and efficient CG simulations of proteins, fast and reliable backmapping remains a challenge. Rule-based methods produce poor all-atom geometries, needing computationally costly refinement through additional simulations. Recently proposed ML approaches outperform traditional baselines but are not transferable between proteins and sometimes generate unphysical atom placements with steric clashes and implausible torsion angles. This work addresses both issues to build a fast, transferable, and reliable generative backmapping tool for CG protein representations. We achieve generalization and reliability through a combined set of innovations: representation based on internal coordinates; an equivariant encoder/prior; a custom loss function that helps ensure local structure, global structure, and physical constraints; and expert curation of high-quality out-of-equilibrium protein data for training. Our results pave the way for out-of-the-box backmapping of coarse-grained simulations for arbitrary proteins.
Direct Estimation of Information Divergence Using Nearest Neighbor Ratios
We propose a direct estimation method for R\'{e}nyi and f-divergence measures based on a new graph theoretical interpretation. Suppose that we are given two sample sets X and Y, respectively with N and M samples, where eta:=M/N is a constant value. Considering the k-nearest neighbor (k-NN) graph of Y in the joint data set (X,Y), we show that the average powered ratio of the number of X points to the number of Y points among all k-NN points is proportional to R\'{e}nyi divergence of X and Y densities. A similar method can also be used to estimate f-divergence measures. We derive bias and variance rates, and show that for the class of gamma-H\"{o}lder smooth functions, the estimator achieves the MSE rate of O(N^{-2gamma/(gamma+d)}). Furthermore, by using a weighted ensemble estimation technique, for density functions with continuous and bounded derivatives of up to the order d, and some extra conditions at the support set boundary, we derive an ensemble estimator that achieves the parametric MSE rate of O(1/N). Our estimators are more computationally tractable than other competing estimators, which makes them appealing in many practical applications.
Feature Programming for Multivariate Time Series Prediction
We introduce the concept of programmable feature engineering for time series modeling and propose a feature programming framework. This framework generates large amounts of predictive features for noisy multivariate time series while allowing users to incorporate their inductive bias with minimal effort. The key motivation of our framework is to view any multivariate time series as a cumulative sum of fine-grained trajectory increments, with each increment governed by a novel spin-gas dynamical Ising model. This fine-grained perspective motivates the development of a parsimonious set of operators that summarize multivariate time series in an abstract fashion, serving as the foundation for large-scale automated feature engineering. Numerically, we validate the efficacy of our method on several synthetic and real-world noisy time series datasets.
Refine Drugs, Don't Complete Them: Uniform-Source Discrete Flows for Fragment-Based Drug Discovery
We introduce InVirtuoGen, a discrete flow generative model for fragmented SMILES for de novo and fragment-constrained generation, and target-property/lead optimization of small molecules. The model learns to transform a uniform source over all possible tokens into the data distribution. Unlike masked models, its training loss accounts for predictions on all sequence positions at every denoising step, shifting the generation paradigm from completion to refinement, and decoupling the number of sampling steps from the sequence length. For de novo generation, InVirtuoGen achieves a stronger quality-diversity pareto frontier than prior fragment-based models and competitive performance on fragment-constrained tasks. For property and lead optimization, we propose a hybrid scheme that combines a genetic algorithm with a Proximal Property Optimization fine-tuning strategy adapted to discrete flows. Our approach sets a new state-of-the-art on the Practical Molecular Optimization benchmark, measured by top-10 AUC across tasks, and yields higher docking scores in lead optimization than previous baselines. InVirtuoGen thus establishes a versatile generative foundation for drug discovery, from early hit finding to multi-objective lead optimization. We further contribute to open science by releasing pretrained checkpoints and code, making our results fully reproduciblehttps://github.com/invirtuolabs/InVirtuoGen_results.



Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks
Molecular Representation Learning (MRL) has proven impactful in numerous biochemical applications such as drug discovery and enzyme design. While Graph Neural Networks (GNNs) are effective at learning molecular representations from a 2D molecular graph or a single 3D structure, existing works often overlook the flexible nature of molecules, which continuously interconvert across conformations via chemical bond rotations and minor vibrational perturbations. To better account for molecular flexibility, some recent works formulate MRL as an ensemble learning problem, focusing on explicitly learning from a set of conformer structures. However, most of these studies have limited datasets, tasks, and models. In this work, we introduce the first MoleculAR Conformer Ensemble Learning (MARCEL) benchmark to thoroughly evaluate the potential of learning on conformer ensembles and suggest promising research directions. MARCEL includes four datasets covering diverse molecule- and reaction-level properties of chemically diverse molecules including organocatalysts and transition-metal catalysts, extending beyond the scope of common GNN benchmarks that are confined to drug-like molecules. In addition, we conduct a comprehensive empirical study, which benchmarks representative 1D, 2D, and 3D molecular representation learning models, along with two strategies that explicitly incorporate conformer ensembles into 3D MRL models. Our findings reveal that direct learning from an accessible conformer space can improve performance on a variety of tasks and models.




Causal de Finetti: On the Identification of Invariant Causal Structure in Exchangeable Data
Learning causal structure from observational data often assumes that we observe independent and identically distributed (i.\,i.\,d) data. The traditional approach aims to find a graphical representation that encodes the same set of conditional independence relationships as those present in the observed distribution. It is known that under i.\,i.\,d assumption, even with infinite data, there is a limit to how fine-grained a causal structure we can identify. To overcome this limitation, recent work has explored using data originating from different, related environments to learn richer causal structure. These approaches implicitly rely on the independent causal mechanisms (ICM) principle, which postulates that the mechanism giving rise to an effect given its causes and the mechanism which generates the causes do not inform or influence each other. Thus, components of the causal model can independently change from environment to environment. Despite its wide application in machine learning and causal inference, there is a lack of statistical formalization of the ICM principle and how it enables identification of richer causal structures from grouped data. Here we present new causal de Finetti theorems which offer a first statistical formalization of ICM principle and show how causal structure identification is possible from exchangeable data. Our work provides theoretical justification for a broad range of techniques leveraging multi-environment data to learn causal structure.
Mean-field underdamped Langevin dynamics and its spacetime discretization
We propose a new method called the N-particle underdamped Langevin algorithm for optimizing a special class of non-linear functionals defined over the space of probability measures. Examples of problems with this formulation include training mean-field neural networks, maximum mean discrepancy minimization and kernel Stein discrepancy minimization. Our algorithm is based on a novel spacetime discretization of the mean-field underdamped Langevin dynamics, for which we provide a new, fast mixing guarantee. In addition, we demonstrate that our algorithm converges globally in total variation distance, bridging the theoretical gap between the dynamics and its practical implementation.
Sample Efficient Reinforcement Learning via Model-Ensemble Exploration and Exploitation
Model-based deep reinforcement learning has achieved success in various domains that require high sample efficiencies, such as Go and robotics. However, there are some remaining issues, such as planning efficient explorations to learn more accurate dynamic models, evaluating the uncertainty of the learned models, and more rational utilization of models. To mitigate these issues, we present MEEE, a model-ensemble method that consists of optimistic exploration and weighted exploitation. During exploration, unlike prior methods directly selecting the optimal action that maximizes the expected accumulative return, our agent first generates a set of action candidates and then seeks out the optimal action that takes both expected return and future observation novelty into account. During exploitation, different discounted weights are assigned to imagined transition tuples according to their model uncertainty respectively, which will prevent model predictive error propagation in agent training. Experiments on several challenging continuous control benchmark tasks demonstrated that our approach outperforms other model-free and model-based state-of-the-art methods, especially in sample complexity.

Embedded Machine Learning for Solar PV Power Regulation in a Remote Microgrid
This paper presents a machine-learning study for solar inverter power regulation in a remote microgrid. Machine learning models for active and reactive power control are respectively trained using an ensemble learning method. Then, unlike conventional schemes that make inferences on a central server in the far-end control center, the proposed scheme deploys the trained models on an embedded edge-computing device near the inverter to reduce the communication delay. Experiments on a real embedded device achieve matched results as on the desktop PC, with about 0.1ms time cost for each inference input.
Real-Time Prediction of Gas Flow Dynamics in Diesel Engines using a Deep Neural Operator Framework
We develop a data-driven deep neural operator framework to approximate multiple output states for a diesel engine and generate real-time predictions with reasonable accuracy. As emission norms become more stringent, the need for fast and accurate models that enable analysis of system behavior have become an essential requirement for system development. The fast transient processes involved in the operation of a combustion engine make it difficult to develop accurate physics-based models for such systems. As an alternative to physics based models, we develop an operator-based regression model (DeepONet) to learn the relevant output states for a mean-value gas flow engine model using the engine operating conditions as input variables. We have adopted a mean-value model as a benchmark for comparison, simulated using Simulink. The developed approach necessitates using the initial conditions of the output states to predict the accurate sequence over the temporal domain. To this end, a sequence-to-sequence approach is embedded into the proposed framework. The accuracy of the model is evaluated by comparing the prediction output to ground truth generated from Simulink model. The maximum mathcal L_2 relative error observed was approximately 6.5%. The sensitivity of the DeepONet model is evaluated under simulated noise conditions and the model shows relatively low sensitivity to noise. The uncertainty in model prediction is further assessed by using a mean ensemble approach. The worst-case error at the (mu + 2sigma) boundary was found to be 12%. The proposed framework provides the ability to predict output states in real-time and enables data-driven learning of complex input-output operator mapping. As a result, this model can be applied during initial development stages, where accurate models may not be available.

Polyatomic Complexes: A topologically-informed learning representation for atomistic systems
Developing robust representations of chemical structures that enable models to learn topological inductive biases is challenging. In this manuscript, we present a representation of atomistic systems. We begin by proving that our representation satisfies all structural, geometric, efficiency, and generalizability constraints. Afterward, we provide a general algorithm to encode any atomistic system. Finally, we report performance comparable to state-of-the-art methods on numerous tasks. We open-source all code and datasets. The code and data are available at https://github.com/rahulkhorana/PolyatomicComplexes.

Pre-trained knowledge elevates large language models beyond traditional chemical reaction optimizers
Modern optimization in experimental chemistry employs algorithmic search through black-box parameter spaces. Here we demonstrate that pre-trained knowledge in large language models (LLMs) fundamentally changes this paradigm. Using six fully enumerated categorical reaction datasets (768 - 5,684 experiments), we benchmark LLM-guided optimization (LLM-GO) against Bayesian optimization (BO) and random sampling. Frontier LLMs consistently match or exceed BO performance across five single-objective datasets, with advantages growing as parameter complexity increases and high-performing conditions become scarce (<5% of space). BO retains superiority only for explicit multi-objective trade-offs. To understand these contrasting behaviors, we introduce a topology-agnostic information theory framework quantifying sampling diversity throughout optimization campaigns. This analysis reveals that LLMs maintain systematically higher exploration entropy than BO across all datasets while achieving superior performance, with advantages most pronounced in solution-scarce parameter spaces where high-entropy exploration typically fails - suggesting that pre-trained domain knowledge enables more effective navigation of chemical parameter space rather than replacing structured exploration strategies. To enable transparent benchmarking and community validation, we release Iron Mind (https://gomes.andrew.cmu.edu/iron-mind), a no-code platform for side-by-side evaluation of human, algorithmic, and LLM optimization campaigns with public leaderboards and complete trajectories. Our findings establish that LLM-GO excels precisely where traditional methods struggle: complex categorical spaces requiring domain understanding rather than mathematical optimization.
The Vendi Score: A Diversity Evaluation Metric for Machine Learning
Diversity is an important criterion for many areas of machine learning (ML), including generative modeling and dataset curation. Yet little work has gone into understanding, formalizing, and measuring diversity in ML. In this paper, we address the diversity evaluation problem by proposing the Vendi Score, which connects and extends ideas from ecology and quantum statistical mechanics to ML. The Vendi Score is defined as the exponential of the Shannon entropy of the eigenvalues of a similarity matrix. This matrix is induced by a user-defined similarity function applied to the sample to be evaluated for diversity. In taking a similarity function as input, the Vendi Score enables its user to specify any desired form of diversity. Importantly, unlike many existing metrics in ML, the Vendi Score doesn't require a reference dataset or distribution over samples or labels, it is therefore general and applicable to any generative model, decoding algorithm, and dataset from any domain where similarity can be defined. We showcased the Vendi Score on molecular generative modeling, a domain where diversity plays an important role in enabling the discovery of novel molecules. We found that the Vendi Score addresses shortcomings of the current diversity metric of choice in that domain. We also applied the Vendi Score to generative models of images and decoding algorithms of text and found it confirms known results about diversity in those domains. Furthermore, we used the Vendi Score to measure mode collapse, a known limitation of generative adversarial networks (GANs). In particular, the Vendi Score revealed that even GANs that capture all the modes of a labeled dataset can be less diverse than the original dataset. Finally, the interpretability of the Vendi Score allowed us to diagnose several benchmark ML datasets for diversity, opening the door for diversity-informed data augmentation.
SEEDS: Emulation of Weather Forecast Ensembles with Diffusion Models
Probabilistic forecasting is crucial to decision-making under uncertainty about future weather. The dominant approach is to use an ensemble of forecasts to represent and quantify uncertainty in operational numerical weather prediction. However, generating ensembles is computationally costly. In this paper, we propose to generate ensemble forecasts at scale by leveraging recent advances in generative artificial intelligence. Our approach learns a data-driven probabilistic diffusion model from the 5-member ensemble GEFS reforecast dataset. The model can then be sampled efficiently to produce realistic weather forecasts, conditioned on a few members of the operational GEFS forecasting system. The generated ensembles have similar predictive skill as the full GEFS 31-member ensemble, evaluated against ERA5 reanalysis, and emulate well the statistics of large physics-based ensembles. We also apply the same methodology to developing a diffusion model for generative post-processing: the model directly learns to correct biases present in the emulated forecasting system by leveraging reanalysis data as labels during training. Ensembles from this generative post-processing model show greater reliability and accuracy, particularly in extreme event classification. In general, they are more reliable and forecast the probability of extreme weather more accurately than the GEFS operational ensemble. Our models achieve these results at less than 1/10th of the computational cost incurred by the operational GEFS system.

Investigating the Pre-Training Dynamics of In-Context Learning: Task Recognition vs. Task Learning
The emergence of in-context learning (ICL) is potentially attributed to two major abilities: task recognition (TR) for recognizing the task from demonstrations and utilizing pre-trained priors, and task learning (TL) for learning from demonstrations. However, relationships between the two abilities and how such relationships affect the emergence of ICL is unclear. In this paper, we take the first step by examining the pre-training dynamics of the emergence of ICL. With carefully designed metrics, we find that these two abilities are, in fact, competitive during pre-training. Moreover, we observe a strong negative correlation between the competition and ICL performance. Further analysis of common pre-training factors (i.e., model size, dataset size, and data curriculum) demonstrates possible ways to manage the competition. Based on these insights, we propose a simple yet effective method to better integrate these two abilities for ICL at inference time. Through adaptive ensemble learning, the performance of ICL can be significantly boosted, enabling two small models to outperform a larger one with more than twice the parameters. The code is available at https://github.com/RUCAIBox/Competitive-ICL.

Diversity of Thought Improves Reasoning Abilities of Large Language Models
Large language models (LLMs) are documented to struggle in settings that require complex reasoning. Nevertheless, instructing the model to break down the problem into smaller reasoning steps (Wei et al., 2022), or ensembling various generations through modifying decoding steps (Wang et al., 2023) boosts performance. Current methods assume that the input prompt is fixed and expect the decoding strategies to introduce the diversity needed for ensembling. In this work, we relax this assumption and discuss how one can create and leverage variations of the input prompt as a means to diversity of thought to improve model performance. We propose a method that automatically improves prompt diversity by soliciting feedback from the LLM to ideate approaches that fit for the problem. We then ensemble the diverse prompts in our method DIV-SE (DIVerse reasoning path Self-Ensemble) across multiple inference calls. We also propose a cost-effective alternative where diverse prompts are used within a single inference call; we call this IDIV-SE (In-call DIVerse reasoning path Self-Ensemble). Under a fixed generation budget, DIV-SE and IDIV-SE outperform the previously discussed baselines using both GPT-3.5 and GPT-4 on several reasoning benchmarks, without modifying the decoding process. Additionally, DIV-SE advances state-of-the-art performance on recent planning benchmarks (Valmeekam et al., 2023), exceeding the highest previously reported accuracy by at least 29.6 percentage points on the most challenging 4/5 Blocksworld task. Our results shed light on how to enforce prompt diversity toward LLM reasoning and thereby improve the pareto frontier of the accuracy-cost trade-off.

Practical and Asymptotically Exact Conditional Sampling in Diffusion Models
Diffusion models have been successful on a range of conditional generation tasks including molecular design and text-to-image generation. However, these achievements have primarily depended on task-specific conditional training or error-prone heuristic approximations. Ideally, a conditional generation method should provide exact samples for a broad range of conditional distributions without requiring task-specific training. To this end, we introduce the Twisted Diffusion Sampler, or TDS. TDS is a sequential Monte Carlo (SMC) algorithm that targets the conditional distributions of diffusion models through simulating a set of weighted particles. The main idea is to use twisting, an SMC technique that enjoys good computational efficiency, to incorporate heuristic approximations without compromising asymptotic exactness. We first find in simulation and in conditional image generation tasks that TDS provides a computational statistical trade-off, yielding more accurate approximations with many particles but with empirical improvements over heuristics with as few as two particles. We then turn to motif-scaffolding, a core task in protein design, using a TDS extension to Riemannian diffusion models. On benchmark test cases, TDS allows flexible conditioning criteria and often outperforms the state of the art.

Achieving Sample and Computational Efficient Reinforcement Learning by Action Space Reduction via Grouping
Reinforcement learning often needs to deal with the exponential growth of states and actions when exploring optimal control in high-dimensional spaces (often known as the curse of dimensionality). In this work, we address this issue by learning the inherent structure of action-wise similar MDP to appropriately balance the performance degradation versus sample/computational complexity. In particular, we partition the action spaces into multiple groups based on the similarity in transition distribution and reward function, and build a linear decomposition model to capture the difference between the intra-group transition kernel and the intra-group rewards. Both our theoretical analysis and experiments reveal a surprising and counter-intuitive result: while a more refined grouping strategy can reduce the approximation error caused by treating actions in the same group as identical, it also leads to increased estimation error when the size of samples or the computation resources is limited. This finding highlights the grouping strategy as a new degree of freedom that can be optimized to minimize the overall performance loss. To address this issue, we formulate a general optimization problem for determining the optimal grouping strategy, which strikes a balance between performance loss and sample/computational complexity. We further propose a computationally efficient method for selecting a nearly-optimal grouping strategy, which maintains its computational complexity independent of the size of the action space.

Foundation Inference Models for Markov Jump Processes
Markov jump processes are continuous-time stochastic processes which describe dynamical systems evolving in discrete state spaces. These processes find wide application in the natural sciences and machine learning, but their inference is known to be far from trivial. In this work we introduce a methodology for zero-shot inference of Markov jump processes (MJPs), on bounded state spaces, from noisy and sparse observations, which consists of two components. First, a broad probability distribution over families of MJPs, as well as over possible observation times and noise mechanisms, with which we simulate a synthetic dataset of hidden MJPs and their noisy observation process. Second, a neural network model that processes subsets of the simulated observations, and that is trained to output the initial condition and rate matrix of the target MJP in a supervised way. We empirically demonstrate that one and the same (pretrained) model can infer, in a zero-shot fashion, hidden MJPs evolving in state spaces of different dimensionalities. Specifically, we infer MJPs which describe (i) discrete flashing ratchet systems, which are a type of Brownian motors, and the conformational dynamics in (ii) molecular simulations, (iii) experimental ion channel data and (iv) simple protein folding models. What is more, we show that our model performs on par with state-of-the-art models which are finetuned to the target datasets.
Internal Causal Mechanisms Robustly Predict Language Model Out-of-Distribution Behaviors
Interpretability research now offers a variety of techniques for identifying abstract internal mechanisms in neural networks. Can such techniques be used to predict how models will behave on out-of-distribution examples? In this work, we provide a positive answer to this question. Through a diverse set of language modeling tasks--including symbol manipulation, knowledge retrieval, and instruction following--we show that the most robust features for correctness prediction are those that play a distinctive causal role in the model's behavior. Specifically, we propose two methods that leverage causal mechanisms to predict the correctness of model outputs: counterfactual simulation (checking whether key causal variables are realized) and value probing (using the values of those variables to make predictions). Both achieve high AUC-ROC in distribution and outperform methods that rely on causal-agnostic features in out-of-distribution settings, where predicting model behaviors is more crucial. Our work thus highlights a novel and significant application for internal causal analysis of language models.

Untangling Gaussian Mixtures
Tangles were originally introduced as a concept to formalize regions of high connectivity in graphs. In recent years, they have also been discovered as a link between structural graph theory and data science: when interpreting similarity in data sets as connectivity between points, finding clusters in the data essentially amounts to finding tangles in the underlying graphs. This paper further explores the potential of tangles in data sets as a means for a formal study of clusters. Real-world data often follow a normal distribution. Accounting for this, we develop a quantitative theory of tangles in data sets drawn from Gaussian mixtures. To this end, we equip the data with a graph structure that models similarity between the points and allows us to apply tangle theory to the data. We provide explicit conditions under which tangles associated with the marginal Gaussian distributions exist asymptotically almost surely. This can be considered as a sufficient formal criterion for the separabability of clusters in the data.
Unraveling the Key Components of OOD Generalization via Diversification
Supervised learning datasets may contain multiple cues that explain the training set equally well, i.e., learning any of them would lead to the correct predictions on the training data. However, many of them can be spurious, i.e., lose their predictive power under a distribution shift and consequently fail to generalize to out-of-distribution (OOD) data. Recently developed "diversification" methods (Lee et al., 2023; Pagliardini et al., 2023) approach this problem by finding multiple diverse hypotheses that rely on different features. This paper aims to study this class of methods and identify the key components contributing to their OOD generalization abilities. We show that (1) diversification methods are highly sensitive to the distribution of the unlabeled data used for diversification and can underperform significantly when away from a method-specific sweet spot. (2) Diversification alone is insufficient for OOD generalization. The choice of the used learning algorithm, e.g., the model's architecture and pretraining, is crucial. In standard experiments (classification on Waterbirds and Office-Home datasets), using the second-best choice leads to an up to 20\% absolute drop in accuracy. (3) The optimal choice of learning algorithm depends on the unlabeled data and vice versa i.e. they are co-dependent. (4) Finally, we show that, in practice, the above pitfalls cannot be alleviated by increasing the number of diverse hypotheses, the major feature of diversification methods. These findings provide a clearer understanding of the critical design factors influencing the OOD generalization abilities of diversification methods. They can guide practitioners in how to use the existing methods best and guide researchers in developing new, better ones.

Feynman-Kac Correctors in Diffusion: Annealing, Guidance, and Product of Experts
While score-based generative models are the model of choice across diverse domains, there are limited tools available for controlling inference-time behavior in a principled manner, e.g. for composing multiple pretrained models. Existing classifier-free guidance methods use a simple heuristic to mix conditional and unconditional scores to approximately sample from conditional distributions. However, such methods do not approximate the intermediate distributions, necessitating additional 'corrector' steps. In this work, we provide an efficient and principled method for sampling from a sequence of annealed, geometric-averaged, or product distributions derived from pretrained score-based models. We derive a weighted simulation scheme which we call Feynman-Kac Correctors (FKCs) based on the celebrated Feynman-Kac formula by carefully accounting for terms in the appropriate partial differential equations (PDEs). To simulate these PDEs, we propose Sequential Monte Carlo (SMC) resampling algorithms that leverage inference-time scaling to improve sampling quality. We empirically demonstrate the utility of our methods by proposing amortized sampling via inference-time temperature annealing, improving multi-objective molecule generation using pretrained models, and improving classifier-free guidance for text-to-image generation. Our code is available at https://github.com/martaskrt/fkc-diffusion.

Anti-Exploration by Random Network Distillation
Despite the success of Random Network Distillation (RND) in various domains, it was shown as not discriminative enough to be used as an uncertainty estimator for penalizing out-of-distribution actions in offline reinforcement learning. In this paper, we revisit these results and show that, with a naive choice of conditioning for the RND prior, it becomes infeasible for the actor to effectively minimize the anti-exploration bonus and discriminativity is not an issue. We show that this limitation can be avoided with conditioning based on Feature-wise Linear Modulation (FiLM), resulting in a simple and efficient ensemble-free algorithm based on Soft Actor-Critic. We evaluate it on the D4RL benchmark, showing that it is capable of achieving performance comparable to ensemble-based methods and outperforming ensemble-free approaches by a wide margin.



Bayesian active learning for optimization and uncertainty quantification in protein docking
Motivation: Ab initio protein docking represents a major challenge for optimizing a noisy and costly "black box"-like function in a high-dimensional space. Despite progress in this field, there is no docking method available for rigorous uncertainty quantification (UQ) of its solution quality (e.g. interface RMSD or iRMSD). Results: We introduce a novel algorithm, Bayesian Active Learning (BAL), for optimization and UQ of such black-box functions and flexible protein docking. BAL directly models the posterior distribution of the global optimum (or native structures for protein docking) with active sampling and posterior estimation iteratively feeding each other. Furthermore, we use complex normal modes to represent a homogeneous Euclidean conformation space suitable for high-dimension optimization and construct funnel-like energy models for encounter complexes. Over a protein docking benchmark set and a CAPRI set including homology docking, we establish that BAL significantly improve against both starting points by rigid docking and refinements by particle swarm optimization, providing for one third targets a top-3 near-native prediction. BAL also generates tight confidence intervals with half range around 25% of iRMSD and confidence level at 85%. Its estimated probability of a prediction being native or not achieves binary classification AUROC at 0.93 and AUPRC over 0.60 (compared to 0.14 by chance); and also found to help ranking predictions. To the best of our knowledge, this study represents the first uncertainty quantification solution for protein docking, with theoretical rigor and comprehensive assessment. Source codes are available at https://github.com/Shen-Lab/BAL.
Full-Atom Peptide Design based on Multi-modal Flow Matching
Peptides, short chains of amino acid residues, play a vital role in numerous biological processes by interacting with other target molecules, offering substantial potential in drug discovery. In this work, we present PepFlow, the first multi-modal deep generative model grounded in the flow-matching framework for the design of full-atom peptides that target specific protein receptors. Drawing inspiration from the crucial roles of residue backbone orientations and side-chain dynamics in protein-peptide interactions, we characterize the peptide structure using rigid backbone frames within the SE(3) manifold and side-chain angles on high-dimensional tori. Furthermore, we represent discrete residue types in the peptide sequence as categorical distributions on the probability simplex. By learning the joint distributions of each modality using derived flows and vector fields on corresponding manifolds, our method excels in the fine-grained design of full-atom peptides. Harnessing the multi-modal paradigm, our approach adeptly tackles various tasks such as fix-backbone sequence design and side-chain packing through partial sampling. Through meticulously crafted experiments, we demonstrate that PepFlow exhibits superior performance in comprehensive benchmarks, highlighting its significant potential in computational peptide design and analysis.
Learning dynamic representations of the functional connectome in neurobiological networks
The static synaptic connectivity of neuronal circuits stands in direct contrast to the dynamics of their function. As in changing community interactions, different neurons can participate actively in various combinations to effect behaviors at different times. We introduce an unsupervised approach to learn the dynamic affinities between neurons in live, behaving animals, and to reveal which communities form among neurons at different times. The inference occurs in two major steps. First, pairwise non-linear affinities between neuronal traces from brain-wide calcium activity are organized by non-negative tensor factorization (NTF). Each factor specifies which groups of neurons are most likely interacting for an inferred interval in time, and for which animals. Finally, a generative model that allows for weighted community detection is applied to the functional motifs produced by NTF to reveal a dynamic functional connectome. Since time codes the different experimental variables (e.g., application of chemical stimuli), this provides an atlas of neural motifs active during separate stages of an experiment (e.g., stimulus application or spontaneous behaviors). Results from our analysis are experimentally validated, confirming that our method is able to robustly predict causal interactions between neurons to generate behavior. Code is available at https://github.com/dyballa/dynamic-connectomes.

Counterfactual Analysis in Dynamic Latent State Models
We provide an optimization-based framework to perform counterfactual analysis in a dynamic model with hidden states. Our framework is grounded in the ``abduction, action, and prediction'' approach to answer counterfactual queries and handles two key challenges where (1) the states are hidden and (2) the model is dynamic. Recognizing the lack of knowledge on the underlying causal mechanism and the possibility of infinitely many such mechanisms, we optimize over this space and compute upper and lower bounds on the counterfactual quantity of interest. Our work brings together ideas from causality, state-space models, simulation, and optimization, and we apply it on a breast cancer case study. To the best of our knowledge, we are the first to compute lower and upper bounds on a counterfactual query in a dynamic latent-state model.
Large-Scale Targeted Cause Discovery with Data-Driven Learning
We propose a novel machine learning approach for inferring causal variables of a target variable from observations. Our focus is on directly inferring a set of causal factors without requiring full causal graph reconstruction, which is computationally challenging in large-scale systems. The identified causal set consists of all potential regulators of the target variable under experimental settings, enabling efficient regulation when intervention costs and feasibility vary across variables. To achieve this, we train a neural network using supervised learning on simulated data to infer causality. By employing a local-inference strategy, our approach scales with linear complexity in the number of variables, efficiently scaling up to thousands of variables. Empirical results demonstrate superior performance in identifying causal relationships within large-scale gene regulatory networks, outperforming existing methods that emphasize full-graph discovery. We validate our model's generalization capability across out-of-distribution graph structures and generating mechanisms, including gene regulatory networks of E. coli and the human K562 cell line. Implementation codes are available at https://github.com/snu-mllab/Targeted-Cause-Discovery.




Gradient-Free Sequential Bayesian Experimental Design via Interacting Particle Systems
We introduce a gradient-free framework for Bayesian Optimal Experimental Design (BOED) in sequential settings, aimed at complex systems where gradient information is unavailable. Our method combines Ensemble Kalman Inversion (EKI) for design optimization with the Affine-Invariant Langevin Dynamics (ALDI) sampler for efficient posterior sampling-both of which are derivative-free and ensemble-based. To address the computational challenges posed by nested expectations in BOED, we propose variational Gaussian and parametrized Laplace approximations that provide tractable upper and lower bounds on the Expected Information Gain (EIG). These approximations enable scalable utility estimation in high-dimensional spaces and PDE-constrained inverse problems. We demonstrate the performance of our framework through numerical experiments ranging from linear Gaussian models to PDE-based inference tasks, highlighting the method's robustness, accuracy, and efficiency in information-driven experimental design.
PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences
The last few years have seen the development of numerous deep learning-based protein-ligand docking methods. They offer huge promise in terms of speed and accuracy. However, despite claims of state-of-the-art performance in terms of crystallographic root-mean-square deviation (RMSD), upon closer inspection, it has become apparent that they often produce physically implausible molecular structures. It is therefore not sufficient to evaluate these methods solely by RMSD to a native binding mode. It is vital, particularly for deep learning-based methods, that they are also evaluated on steric and energetic criteria. We present PoseBusters, a Python package that performs a series of standard quality checks using the well-established cheminformatics toolkit RDKit. Only methods that both pass these checks and predict native-like binding modes should be classed as having "state-of-the-art" performance. We use PoseBusters to compare five deep learning-based docking methods (DeepDock, DiffDock, EquiBind, TankBind, and Uni-Mol) and two well-established standard docking methods (AutoDock Vina and CCDC Gold) with and without an additional post-prediction energy minimisation step using a molecular mechanics force field. We show that both in terms of physical plausibility and the ability to generalise to examples that are distinct from the training data, no deep learning-based method yet outperforms classical docking tools. In addition, we find that molecular mechanics force fields contain docking-relevant physics missing from deep-learning methods. PoseBusters allows practitioners to assess docking and molecular generation methods and may inspire new inductive biases still required to improve deep learning-based methods, which will help drive the development of more accurate and more realistic predictions.
Diverse Projection Ensembles for Distributional Reinforcement Learning
In contrast to classical reinforcement learning, distributional reinforcement learning algorithms aim to learn the distribution of returns rather than their expected value. Since the nature of the return distribution is generally unknown a priori or arbitrarily complex, a common approach finds approximations within a set of representable, parametric distributions. Typically, this involves a projection of the unconstrained distribution onto the set of simplified distributions. We argue that this projection step entails a strong inductive bias when coupled with neural networks and gradient descent, thereby profoundly impacting the generalization behavior of learned models. In order to facilitate reliable uncertainty estimation through diversity, this work studies the combination of several different projections and representations in a distributional ensemble. We establish theoretical properties of such projection ensembles and derive an algorithm that uses ensemble disagreement, measured by the average 1-Wasserstein distance, as a bonus for deep exploration. We evaluate our algorithm on the behavior suite benchmark and find that diverse projection ensembles lead to significant performance improvements over existing methods on a wide variety of tasks with the most pronounced gains in directed exploration problems.
Deep Unsupervised Learning using Nonequilibrium Thermodynamics
A central problem in machine learning involves modeling complex data-sets using highly flexible families of probability distributions in which learning, sampling, inference, and evaluation are still analytically or computationally tractable. Here, we develop an approach that simultaneously achieves both flexibility and tractability. The essential idea, inspired by non-equilibrium statistical physics, is to systematically and slowly destroy structure in a data distribution through an iterative forward diffusion process. We then learn a reverse diffusion process that restores structure in data, yielding a highly flexible and tractable generative model of the data. This approach allows us to rapidly learn, sample from, and evaluate probabilities in deep generative models with thousands of layers or time steps, as well as to compute conditional and posterior probabilities under the learned model. We additionally release an open source reference implementation of the algorithm.

On Computing Optimal Tree Ensembles
Random forests and, more generally, (decision\nobreakdash-)tree ensembles are widely used methods for classification and regression. Recent algorithmic advances allow to compute decision trees that are optimal for various measures such as their size or depth. We are not aware of such research for tree ensembles and aim to contribute to this area. Mainly, we provide two novel algorithms and corresponding lower bounds. First, we are able to carry over and substantially improve on tractability results for decision trees, obtaining a (6delta D S)^S cdot poly-time algorithm, where S is the number of cuts in the tree ensemble, D the largest domain size, and delta is the largest number of features in which two examples differ. To achieve this, we introduce the witness-tree technique which also seems promising for practice. Second, we show that dynamic programming, which has been successful for decision trees, may also be viable for tree ensembles, providing an ell^n cdot poly-time algorithm, where ell is the number of trees and n the number of examples. Finally, we compare the number of cuts necessary to classify training data sets for decision trees and tree ensembles, showing that ensembles may need exponentially fewer cuts for increasing number of trees.
PEFT for Speech: Unveiling Optimal Placement, Merging Strategies, and Ensemble Techniques
Parameter-Efficient Fine-Tuning (PEFT) is increasingly recognized as an effective method in speech processing. However, the optimal approach and the placement of PEFT methods remain inconclusive. Our study conducts extensive experiments to compare different PEFT methods and their layer-wise placement adapting Differentiable Architecture Search (DARTS). We also explore the use of ensemble learning to leverage diverse PEFT strategies. The results reveal that DARTS does not outperform the baseline approach, which involves inserting the same PEFT method into all layers of a Self-Supervised Learning (SSL) model. In contrast, an ensemble learning approach, particularly one employing majority voting, demonstrates superior performance. Our statistical evidence indicates that different PEFT methods learn in varied ways. This variation might explain why the synergistic integration of various PEFT methods through ensemble learning can harness their unique learning capabilities more effectively compared to individual layer-wise optimization.

Generative Discovery of Novel Chemical Designs using Diffusion Modeling and Transformer Deep Neural Networks with Application to Deep Eutectic Solvents
We report a series of deep learning models to solve complex forward and inverse design problems in molecular modeling and design. Using both diffusion models inspired by nonequilibrium thermodynamics and attention-based transformer architectures, we demonstrate a flexible framework to capture complex chemical structures. First trained on the QM9 dataset and a series of quantum mechanical properties (e.g. homo, lumo, free energy, heat capacity, etc.), we then generalize the model to study and design key properties of deep eutectic solvents. In addition to separate forward and inverse models, we also report an integrated fully prompt-based multi-task generative pretrained transformer model that solves multiple forward, inverse design, and prediction tasks, flexibly and within one model. We show that the multi-task generative model has the overall best performance and allows for flexible integration of multiple objectives, within one model, and for distinct chemistries, suggesting that synergies emerge during training of this large language model. Trained jointly in tasks related to the QM9 dataset and deep eutectic solvents (DESs), the model can predict various quantum mechanical properties and critical properties to achieve deep eutectic solvent behavior. Several novel combinations of DESs are proposed based on this framework.
Multimarginal generative modeling with stochastic interpolants
Given a set of K probability densities, we consider the multimarginal generative modeling problem of learning a joint distribution that recovers these densities as marginals. The structure of this joint distribution should identify multi-way correspondences among the prescribed marginals. We formalize an approach to this task within a generalization of the stochastic interpolant framework, leading to efficient learning algorithms built upon dynamical transport of measure. Our generative models are defined by velocity and score fields that can be characterized as the minimizers of simple quadratic objectives, and they are defined on a simplex that generalizes the time variable in the usual dynamical transport framework. The resulting transport on the simplex is influenced by all marginals, and we show that multi-way correspondences can be extracted. The identification of such correspondences has applications to style transfer, algorithmic fairness, and data decorruption. In addition, the multimarginal perspective enables an efficient algorithm for reducing the dynamical transport cost in the ordinary two-marginal setting. We demonstrate these capacities with several numerical examples.

Learning Collective Variables for Protein Folding with Labeled Data Augmentation through Geodesic Interpolation
In molecular dynamics (MD) simulations, rare events, such as protein folding, are typically studied by means of enhanced sampling techniques, most of which rely on the definition of a collective variable (CV) along which the acceleration occurs. Obtaining an expressive CV is crucial, but often hindered by the lack of information about the particular event, e.g., the transition from unfolded to folded conformation. We propose a simulation-free data augmentation strategy using physics-inspired metrics to generate geodesic interpolations resembling protein folding transitions, thereby improving sampling efficiency without true transition state samples. Leveraging interpolation progress parameters, we introduce a regression-based learning scheme for CV models, which outperforms classifier-based methods when transition state data is limited and noisy
Kolmogorov--Arnold networks in molecular dynamics
We explore the integration of Kolmogorov Networks (KANs) into molecular dynamics (MD) simulations to improve interatomic potentials. We propose that widely used potentials, such as the Lennard-Jones (LJ) potential, the embedded atom model (EAM), and artificial neural network (ANN) potentials, can be interpreted within the KAN framework. Specifically, we demonstrate that the descriptors for ANN potentials, typically constructed using polynomials, can be redefined using KAN's non-linear functions. By employing linear or cubic spline interpolations for these KAN functions, we show that the computational cost of evaluating ANN potentials and their derivatives is reduced.
Value Function is All You Need: A Unified Learning Framework for Ride Hailing Platforms
Large ride-hailing platforms, such as DiDi, Uber and Lyft, connect tens of thousands of vehicles in a city to millions of ride demands throughout the day, providing great promises for improving transportation efficiency through the tasks of order dispatching and vehicle repositioning. Existing studies, however, usually consider the two tasks in simplified settings that hardly address the complex interactions between the two, the real-time fluctuations between supply and demand, and the necessary coordinations due to the large-scale nature of the problem. In this paper we propose a unified value-based dynamic learning framework (V1D3) for tackling both tasks. At the center of the framework is a globally shared value function that is updated continuously using online experiences generated from real-time platform transactions. To improve the sample-efficiency and the robustness, we further propose a novel periodic ensemble method combining the fast online learning with a large-scale offline training scheme that leverages the abundant historical driver trajectory data. This allows the proposed framework to adapt quickly to the highly dynamic environment, to generalize robustly to recurrent patterns and to drive implicit coordinations among the population of managed vehicles. Extensive experiments based on real-world datasets show considerably improvements over other recently proposed methods on both tasks. Particularly, V1D3 outperforms the first prize winners of both dispatching and repositioning tracks in the KDD Cup 2020 RL competition, achieving state-of-the-art results on improving both total driver income and user experience related metrics.
HyPINO: Multi-Physics Neural Operators via HyperPINNs and the Method of Manufactured Solutions
We present HyPINO, a multi-physics neural operator designed for zero-shot generalization across a broad class of parametric PDEs without requiring task-specific fine-tuning. Our approach combines a Swin Transformer-based hypernetwork with mixed supervision: (i) labeled data from analytical solutions generated via the Method of Manufactured Solutions (MMS), and (ii) unlabeled samples optimized using physics-informed objectives. The model maps PDE parametrizations to target Physics-Informed Neural Networks (PINNs) and can handle linear elliptic, hyperbolic, and parabolic equations in two dimensions with varying source terms, geometries, and mixed Dirichlet/Neumann boundary conditions, including interior boundaries. HyPINO achieves strong zero-shot accuracy on seven benchmark problems from PINN literature, outperforming U-Nets, Poseidon, and Physics-Informed Neural Operators (PINO). Further, we introduce an iterative refinement procedure that compares the physics of the generated PINN to the requested PDE and uses the discrepancy to generate a "delta" PINN. Summing their contributions and repeating this process forms an ensemble whose combined solution progressively reduces the error on six benchmarks and achieves over 100x gain in average L_2 loss in the best case, while retaining forward-only inference. Additionally, we evaluate the fine-tuning behavior of PINNs initialized by HyPINO and show that they converge faster and to lower final error than both randomly initialized and Reptile-meta-learned PINNs on five benchmarks, performing on par on the remaining two. Our results highlight the potential of this scalable approach as a foundation for extending neural operators toward solving increasingly complex, nonlinear, and high-dimensional PDE problems with significantly improved accuracy and reduced computational cost.
Intrinsic Sliced Wasserstein Distances for Comparing Collections of Probability Distributions on Manifolds and Graphs
Collections of probability distributions arise in a variety of applications ranging from user activity pattern analysis to brain connectomics. In practice these distributions can be defined over diverse domain types including finite intervals, circles, cylinders, spheres, other manifolds, and graphs. This paper introduces an approach for detecting differences between two collections of distributions over such general domains. To this end, we propose the intrinsic slicing construction that yields a novel class of Wasserstein distances on manifolds and graphs. These distances are Hilbert embeddable, allowing us to reduce the distribution collection comparison problem to a more familiar mean testing problem in a Hilbert space. We provide two testing procedures one based on resampling and another on combining p-values from coordinate-wise tests. Our experiments in various synthetic and real data settings show that the resulting tests are powerful and the p-values are well-calibrated.

Dense Hebbian neural networks: a replica symmetric picture of supervised learning
We consider dense, associative neural-networks trained by a teacher (i.e., with supervision) and we investigate their computational capabilities analytically, via statistical-mechanics of spin glasses, and numerically, via Monte Carlo simulations. In particular, we obtain a phase diagram summarizing their performance as a function of the control parameters such as quality and quantity of the training dataset, network storage and noise, that is valid in the limit of large network size and structureless datasets: these networks may work in a ultra-storage regime (where they can handle a huge amount of patterns, if compared with shallow neural networks) or in a ultra-detection regime (where they can perform pattern recognition at prohibitive signal-to-noise ratios, if compared with shallow neural networks). Guided by the random theory as a reference framework, we also test numerically learning, storing and retrieval capabilities shown by these networks on structured datasets as MNist and Fashion MNist. As technical remarks, from the analytic side, we implement large deviations and stability analysis within Guerra's interpolation to tackle the not-Gaussian distributions involved in the post-synaptic potentials while, from the computational counterpart, we insert Plefka approximation in the Monte Carlo scheme, to speed up the evaluation of the synaptic tensors, overall obtaining a novel and broad approach to investigate supervised learning in neural networks, beyond the shallow limit, in general.

Bayesian Hierarchical Models for Quantitative Estimates for Performance metrics applied to Saddle Search Algorithms
Rigorous performance evaluation is essential for developing robust algorithms for high-throughput computational chemistry. Traditional benchmarking, however, often struggles to account for system-specific variability, making it difficult to form actionable conclusions. We present a Bayesian hierarchical modeling framework that rigorously quantifies performance metrics and their uncertainty, enabling a nuanced comparison of algorithmic strategies. We apply this framework to analyze the Dimer method, comparing Conjugate Gradient (CG) and L-BFGS rotation optimizers, with and without the removal of external rotations, across a benchmark of 500 molecular systems. Our analysis confirms that CG offers higher overall robustness than L-BFGS in this context. While the theoretically-motivated removal of external rotations led to higher computational cost (>40% more energy and force calls) for most systems in this set, our models also reveal a subtle interplay, hinting that this feature may improve the reliability of the L-BFGS optimizer. Rather than identifying a single superior method, our findings support the design of adaptive "chain of methods" workflows. This work showcases how a robust statistical paradigm can move beyond simple performance rankings to inform the intelligent, context-dependent application of computational chemistry methods.

Chaos as an interpretable benchmark for forecasting and data-driven modelling
The striking fractal geometry of strange attractors underscores the generative nature of chaos: like probability distributions, chaotic systems can be repeatedly measured to produce arbitrarily-detailed information about the underlying attractor. Chaotic systems thus pose a unique challenge to modern statistical learning techniques, while retaining quantifiable mathematical properties that make them controllable and interpretable as benchmarks. Here, we present a growing database currently comprising 131 known chaotic dynamical systems spanning fields such as astrophysics, climatology, and biochemistry. Each system is paired with precomputed multivariate and univariate time series. Our dataset has comparable scale to existing static time series databases; however, our systems can be re-integrated to produce additional datasets of arbitrary length and granularity. Our dataset is annotated with known mathematical properties of each system, and we perform feature analysis to broadly categorize the diverse dynamics present across the collection. Chaotic systems inherently challenge forecasting models, and across extensive benchmarks we correlate forecasting performance with the degree of chaos present. We also exploit the unique generative properties of our dataset in several proof-of-concept experiments: surrogate transfer learning to improve time series classification, importance sampling to accelerate model training, and benchmarking symbolic regression algorithms.
Interpretability in the Wild: a Circuit for Indirect Object Identification in GPT-2 small
Research in mechanistic interpretability seeks to explain behaviors of machine learning models in terms of their internal components. However, most previous work either focuses on simple behaviors in small models, or describes complicated behaviors in larger models with broad strokes. In this work, we bridge this gap by presenting an explanation for how GPT-2 small performs a natural language task called indirect object identification (IOI). Our explanation encompasses 26 attention heads grouped into 7 main classes, which we discovered using a combination of interpretability approaches relying on causal interventions. To our knowledge, this investigation is the largest end-to-end attempt at reverse-engineering a natural behavior "in the wild" in a language model. We evaluate the reliability of our explanation using three quantitative criteria--faithfulness, completeness and minimality. Though these criteria support our explanation, they also point to remaining gaps in our understanding. Our work provides evidence that a mechanistic understanding of large ML models is feasible, opening opportunities to scale our understanding to both larger models and more complex tasks.


Controlling Ensemble Variance in Diffusion Models: An Application for Reanalyses Downscaling
In recent years, diffusion models have emerged as powerful tools for generating ensemble members in meteorology. In this work, we demonstrate that a Denoising Diffusion Implicit Model (DDIM) can effectively control ensemble variance by varying the number of diffusion steps. Introducing a theoretical framework, we relate diffusion steps to the variance expressed by the reverse diffusion process. Focusing on reanalysis downscaling, we propose an ensemble diffusion model for the full ERA5-to-CERRA domain, generating variance-calibrated ensemble members for wind speed at full spatial and temporal resolution. Our method aligns global mean variance with a reference ensemble dataset and ensures spatial variance is distributed in accordance with observed meteorological variability. Additionally, we address the lack of ensemble information in the CARRA dataset, showcasing the utility of our approach for efficient, high-resolution ensemble generation.

HMC with Normalizing Flows
We propose using Normalizing Flows as a trainable kernel within the molecular dynamics update of Hamiltonian Monte Carlo (HMC). By learning (invertible) transformations that simplify our dynamics, we can outperform traditional methods at generating independent configurations. We show that, using a carefully constructed network architecture, our approach can be easily scaled to large lattice volumes with minimal retraining effort. The source code for our implementation is publicly available online at https://github.com/nftqcd/fthmc.

PANTHER: Pathway Augmented Nonnegative Tensor factorization for HighER-order feature learning
Genetic pathways usually encode molecular mechanisms that can inform targeted interventions. It is often challenging for existing machine learning approaches to jointly model genetic pathways (higher-order features) and variants (atomic features), and present to clinicians interpretable models. In order to build more accurate and better interpretable machine learning models for genetic medicine, we introduce Pathway Augmented Nonnegative Tensor factorization for HighER-order feature learning (PANTHER). PANTHER selects informative genetic pathways that directly encode molecular mechanisms. We apply genetically motivated constrained tensor factorization to group pathways in a way that reflects molecular mechanism interactions. We then train a softmax classifier for disease types using the identified pathway groups. We evaluated PANTHER against multiple state-of-the-art constrained tensor/matrix factorization models, as well as group guided and Bayesian hierarchical models. PANTHER outperforms all state-of-the-art comparison models significantly (p<0.05). Our experiments on large scale Next Generation Sequencing (NGS) and whole-genome genotyping datasets also demonstrated wide applicability of PANTHER. We performed feature analysis in predicting disease types, which suggested insights and benefits of the identified pathway groups.
Regularized Langevin Dynamics for Combinatorial Optimization
This work proposes a simple yet effective sampling framework for combinatorial optimization (CO). Our method builds on discrete Langevin dynamics (LD), an efficient gradient-guided generative paradigm. However, we observe that directly applying LD often leads to limited exploration. To overcome this limitation, we propose the Regularized Langevin Dynamics (RLD), which enforces an expected distance between the sampled and current solutions, effectively avoiding local minima. We develop two CO solvers on top of RLD, one based on simulated annealing (SA), and the other one based on neural network (NN). Empirical results on three classic CO problems demonstrate that both of our methods can achieve comparable or better performance against the previous state-of-the-art (SOTA) SA- and NN-based solvers. In particular, our SA algorithm reduces the runtime of the previous SOTA SA method by up to 80\%, while achieving equal or superior performance. In summary, RLD offers a promising framework for enhancing both traditional heuristics and NN models to solve CO problems. Our code is available at https://github.com/Shengyu-Feng/RLD4CO.
Constructor Theory of Thermodynamics
All current formulations of thermodynamics invoke some form of coarse-graining or ensembles as the supposed link between their own laws and the microscopic laws of motion. They deal only with ensemble-averages, expectation values, macroscopic limits, infinite heat baths, etc., not with the details of physical variables of individual microscopic systems. They are consistent with the laws of motion for finite systems only in certain approximations, which improve with increasing scale, given various assumptions about initial conditions which are neither specified precisely nor even thought to hold exactly in nature. Here I propose a new formulation of the zeroth, first and second laws, improving upon the axiomatic approach to thermodynamics (Carath\'eodory, 1909; Lieb & Yngvason, 1999), via the principles of the recently proposed constructor theory. Specifically, I provide a non-approximative, scale-independent formulation of 'adiabatic accessibility'; this in turn provides a non-approximative, scale-independent distinction between work and heat and reveals an unexpected connection between information theory and the first law of thermodynamics (not just the second). It also achieves the long-sought unification of the axiomatic approach with Kelvin's.
Dense Hebbian neural networks: a replica symmetric picture of unsupervised learning
We consider dense, associative neural-networks trained with no supervision and we investigate their computational capabilities analytically, via a statistical-mechanics approach, and numerically, via Monte Carlo simulations. In particular, we obtain a phase diagram summarizing their performance as a function of the control parameters such as the quality and quantity of the training dataset and the network storage, valid in the limit of large network size and structureless datasets. Moreover, we establish a bridge between macroscopic observables standardly used in statistical mechanics and loss functions typically used in the machine learning. As technical remarks, from the analytic side, we implement large deviations and stability analysis within Guerra's interpolation to tackle the not-Gaussian distributions involved in the post-synaptic potentials while, from the computational counterpart, we insert Plefka approximation in the Monte Carlo scheme, to speed up the evaluation of the synaptic tensors, overall obtaining a novel and broad approach to investigate neural networks in general.
Diffusion Twigs with Loop Guidance for Conditional Graph Generation
We introduce a novel score-based diffusion framework named Twigs that incorporates multiple co-evolving flows for enriching conditional generation tasks. Specifically, a central or trunk diffusion process is associated with a primary variable (e.g., graph structure), and additional offshoot or stem processes are dedicated to dependent variables (e.g., graph properties or labels). A new strategy, which we call loop guidance, effectively orchestrates the flow of information between the trunk and the stem processes during sampling. This approach allows us to uncover intricate interactions and dependencies, and unlock new generative capabilities. We provide extensive experiments to demonstrate strong performance gains of the proposed method over contemporary baselines in the context of conditional graph generation, underscoring the potential of Twigs in challenging generative tasks such as inverse molecular design and molecular optimization.
Temperature Steerable Flows and Boltzmann Generators
Boltzmann generators approach the sampling problem in many-body physics by combining a normalizing flow and a statistical reweighting method to generate samples in thermodynamic equilibrium. The equilibrium distribution is usually defined by an energy function and a thermodynamic state. Here we propose temperature-steerable flows (TSF) which are able to generate a family of probability densities parametrized by a choosable temperature parameter. TSFs can be embedded in generalized ensemble sampling frameworks to sample a physical system across multiple thermodynamic states.
Randomized Ensembled Double Q-Learning: Learning Fast Without a Model
Using a high Update-To-Data (UTD) ratio, model-based methods have recently achieved much higher sample efficiency than previous model-free methods for continuous-action DRL benchmarks. In this paper, we introduce a simple model-free algorithm, Randomized Ensembled Double Q-Learning (REDQ), and show that its performance is just as good as, if not better than, a state-of-the-art model-based algorithm for the MuJoCo benchmark. Moreover, REDQ can achieve this performance using fewer parameters than the model-based method, and with less wall-clock run time. REDQ has three carefully integrated ingredients which allow it to achieve its high performance: (i) a UTD ratio >> 1; (ii) an ensemble of Q functions; (iii) in-target minimization across a random subset of Q functions from the ensemble. Through carefully designed experiments, we provide a detailed analysis of REDQ and related model-free algorithms. To our knowledge, REDQ is the first successful model-free DRL algorithm for continuous-action spaces using a UTD ratio >> 1.
Examining the Source of Defects from a Mechanical Perspective for 3D Anomaly Detection
In this paper, we explore a novel approach to 3D anomaly detection (AD) that goes beyond merely identifying anomalies based on structural characteristics. Our primary perspective is that most anomalies arise from unpredictable defective forces originating from both internal and external sources. To address these anomalies, we seek out opposing forces that can help correct them. Therefore, we introduce the Mechanics Complementary Model-based Framework for the 3D-AD task (MC4AD), which generates internal and external corrective forces for each point. We first propose a Diverse Anomaly-Generation (DA-Gen) module designed to simulate various types of anomalies. Next, we present the Corrective Force Prediction Network (CFP-Net), which uses complementary representations for point-level analysis to simulate the different contributions from internal and external corrective forces. To ensure the corrective forces are constrained effectively, we have developed a combined loss function that includes a new symmetric loss and an overall loss. Notably, we implement a Hierarchical Quality Control (HQC) strategy based on a three-way decision process and contribute a dataset titled Anomaly-IntraVariance, which incorporates intraclass variance to evaluate our model. As a result, the proposed MC4AD has been proven effective through theory and experimentation. The experimental results demonstrate that our approach yields nine state-of-the-art performances, achieving optimal results with minimal parameters and the fastest inference speed across five existing datasets, in addition to the proposed Anomaly-IntraVariance dataset. The source is available at https://github.com/hzzzzzhappy/MC4AD


Skillful joint probabilistic weather forecasting from marginals
Machine learning (ML)-based weather models have rapidly risen to prominence due to their greater accuracy and speed than traditional forecasts based on numerical weather prediction (NWP), recently outperforming traditional ensembles in global probabilistic weather forecasting. This paper presents FGN, a simple, scalable and flexible modeling approach which significantly outperforms the current state-of-the-art models. FGN generates ensembles via learned model-perturbations with an ensemble of appropriately constrained models. It is trained directly to minimize the continuous rank probability score (CRPS) of per-location forecasts. It produces state-of-the-art ensemble forecasts as measured by a range of deterministic and probabilistic metrics, makes skillful ensemble tropical cyclone track predictions, and captures joint spatial structure despite being trained only on marginals.
Chemical classification program synthesis using generative artificial intelligence
Accurately classifying chemical structures is essential for cheminformatics and bioinformatics, including tasks such as identifying bioactive compounds of interest, screening molecules for toxicity to humans, finding non-organic compounds with desirable material properties, or organizing large chemical libraries for drug discovery or environmental monitoring. However, manual classification is labor-intensive and difficult to scale to large chemical databases. Existing automated approaches either rely on manually constructed classification rules, or the use of deep learning methods that lack explainability. This work presents an approach that uses generative artificial intelligence to automatically write chemical classifier programs for classes in the Chemical Entities of Biological Interest (ChEBI) database. These programs can be used for efficient deterministic run-time classification of SMILES structures, with natural language explanations. The programs themselves constitute an explainable computable ontological model of chemical class nomenclature, which we call the ChEBI Chemical Class Program Ontology (C3PO). We validated our approach against the ChEBI database, and compared our results against state of the art deep learning models. We also demonstrate the use of C3PO to classify out-of-distribution examples taken from metabolomics repositories and natural product databases. We also demonstrate the potential use of our approach to find systematic classification errors in existing chemical databases, and show how an ensemble artificial intelligence approach combining generated ontologies, automated literature search, and multimodal vision models can be used to pinpoint potential errors requiring expert validation
A Neural Scaling Law from Lottery Ticket Ensembling
Neural scaling laws (NSL) refer to the phenomenon where model performance improves with scale. Sharma & Kaplan analyzed NSL using approximation theory and predict that MSE losses decay as N^{-alpha}, alpha=4/d, where N is the number of model parameters, and d is the intrinsic input dimension. Although their theory works well for some cases (e.g., ReLU networks), we surprisingly find that a simple 1D problem y=x^2 manifests a different scaling law (alpha=1) from their predictions (alpha=4). We opened the neural networks and found that the new scaling law originates from lottery ticket ensembling: a wider network on average has more "lottery tickets", which are ensembled to reduce the variance of outputs. We support the ensembling mechanism by mechanistically interpreting single neural networks, as well as studying them statistically. We attribute the N^{-1} scaling law to the "central limit theorem" of lottery tickets. Finally, we discuss its potential implications for large language models and statistical physics-type theories of learning.

From Neurons to Neutrons: A Case Study in Interpretability
Mechanistic Interpretability (MI) promises a path toward fully understanding how neural networks make their predictions. Prior work demonstrates that even when trained to perform simple arithmetic, models can implement a variety of algorithms (sometimes concurrently) depending on initialization and hyperparameters. Does this mean neuron-level interpretability techniques have limited applicability? We argue that high-dimensional neural networks can learn low-dimensional representations of their training data that are useful beyond simply making good predictions. Such representations can be understood through the mechanistic interpretability lens and provide insights that are surprisingly faithful to human-derived domain knowledge. This indicates that such approaches to interpretability can be useful for deriving a new understanding of a problem from models trained to solve it. As a case study, we extract nuclear physics concepts by studying models trained to reproduce nuclear data.

RetroBridge: Modeling Retrosynthesis with Markov Bridges
Retrosynthesis planning is a fundamental challenge in chemistry which aims at designing reaction pathways from commercially available starting materials to a target molecule. Each step in multi-step retrosynthesis planning requires accurate prediction of possible precursor molecules given the target molecule and confidence estimates to guide heuristic search algorithms. We model single-step retrosynthesis planning as a distribution learning problem in a discrete state space. First, we introduce the Markov Bridge Model, a generative framework aimed to approximate the dependency between two intractable discrete distributions accessible via a finite sample of coupled data points. Our framework is based on the concept of a Markov bridge, a Markov process pinned at its endpoints. Unlike diffusion-based methods, our Markov Bridge Model does not need a tractable noise distribution as a sampling proxy and directly operates on the input product molecules as samples from the intractable prior distribution. We then address the retrosynthesis planning problem with our novel framework and introduce RetroBridge, a template-free retrosynthesis modeling approach that achieves state-of-the-art results on standard evaluation benchmarks.
Learning Iterative Reasoning through Energy Diffusion
We introduce iterative reasoning through energy diffusion (IRED), a novel framework for learning to reason for a variety of tasks by formulating reasoning and decision-making problems with energy-based optimization. IRED learns energy functions to represent the constraints between input conditions and desired outputs. After training, IRED adapts the number of optimization steps during inference based on problem difficulty, enabling it to solve problems outside its training distribution -- such as more complex Sudoku puzzles, matrix completion with large value magnitudes, and pathfinding in larger graphs. Key to our method's success is two novel techniques: learning a sequence of annealed energy landscapes for easier inference and a combination of score function and energy landscape supervision for faster and more stable training. Our experiments show that IRED outperforms existing methods in continuous-space reasoning, discrete-space reasoning, and planning tasks, particularly in more challenging scenarios. Code and visualizations at https://energy-based-model.github.io/ired/

From thermodynamics to protein design: Diffusion models for biomolecule generation towards autonomous protein engineering
Protein design with desirable properties has been a significant challenge for many decades. Generative artificial intelligence is a promising approach and has achieved great success in various protein generation tasks. Notably, diffusion models stand out for their robust mathematical foundations and impressive generative capabilities, offering unique advantages in certain applications such as protein design. In this review, we first give the definition and characteristics of diffusion models and then focus on two strategies: Denoising Diffusion Probabilistic Models and Score-based Generative Models, where DDPM is the discrete form of SGM. Furthermore, we discuss their applications in protein design, peptide generation, drug discovery, and protein-ligand interaction. Finally, we outline the future perspectives of diffusion models to advance autonomous protein design and engineering. The E(3) group consists of all rotations, reflections, and translations in three-dimensions. The equivariance on the E(3) group can keep the physical stability of the frame of each amino acid as much as possible, and we reflect on how to keep the diffusion model E(3) equivariant for protein generation.
PepTune: De Novo Generation of Therapeutic Peptides with Multi-Objective-Guided Discrete Diffusion
Peptide therapeutics, a major class of medicines, have achieved remarkable success across diseases such as diabetes and cancer, with landmark examples such as GLP-1 receptor agonists revolutionizing the treatment of type-2 diabetes and obesity. Despite their success, designing peptides that satisfy multiple conflicting objectives, such as target binding affinity, solubility, and membrane permeability, remains a major challenge. Classical drug development and structure-based design are ineffective for such tasks, as they fail to optimize global functional properties critical for therapeutic efficacy. Existing generative frameworks are largely limited to continuous spaces, unconditioned outputs, or single-objective guidance, making them unsuitable for discrete sequence optimization across multiple properties. To address this, we present PepTune, a multi-objective discrete diffusion model for the simultaneous generation and optimization of therapeutic peptide SMILES. Built on the Masked Discrete Language Model (MDLM) framework, PepTune ensures valid peptide structures with state-dependent masking schedules and penalty-based objectives. To guide the diffusion process, we propose a Monte Carlo Tree Search (MCTS)-based strategy that balances exploration and exploitation to iteratively refine Pareto-optimal sequences. MCTS integrates classifier-based rewards with search-tree expansion, overcoming gradient estimation challenges and data sparsity inherent to discrete spaces. Using PepTune, we generate diverse, chemically-modified peptides optimized for multiple therapeutic properties, including target binding affinity, membrane permeability, solubility, hemolysis, and non-fouling characteristics on various disease-relevant targets. In total, our results demonstrate that MCTS-guided discrete diffusion is a powerful and modular approach for multi-objective sequence design in discrete state spaces.


Mitigating Premature Exploitation in Particle-based Monte Carlo for Inference-Time Scaling
Inference-Time Scaling (ITS) improves language models by allocating more computation at generation time. Particle Filtering (PF) has emerged as a strong ITS method for complex mathematical reasoning tasks, but it is vulnerable when guided by process reward models, which often assign overconfident scores early in the reasoning process. This causes PF to suffer from premature exploitation: it myopically commits to locally promising trajectories, prunes potentially correct hypotheses, and converges to suboptimal solutions. This failure mode, known as particle impoverishment, is especially severe under constrained computational budgets. To address this, we analyze the problem and identify two root causes: a lack of diversity in the particle set due to overconfident resampling and consequent inability to assess the potential of a reasoning path. We introduce Entropic Particle Filtering (ePF), an algorithm that integrates two new techniques to solve these issues. The first technique, Entropic Annealing (EA), directly mitigates particle impoverishment by monitoring search diversity via entropy; when diversity drops, it intervenes by dynamically annealing the resampling distribution to preserve exploration. The second, an enhancement called Look-ahead Modulation (LaM), adds a predictive guide to evaluate a state's potential based on its successors. On several challenging math benchmarks, ePF significantly outperforms strong baselines and achieves up to a 50 % relative improvement in task reward. Together, these methods improve PF's resilience by balancing the exploration of diverse solution spaces with the exploitation of high-reward regions, ultimately leading to higher-quality solutions.

A Bayesian Flow Network Framework for Chemistry Tasks
In this work, we introduce ChemBFN, a language model that handles chemistry tasks based on Bayesian flow networks working on discrete data. A new accuracy schedule is proposed to improve the sampling quality by significantly reducing the reconstruction loss. We show evidence that our method is appropriate for generating molecules with satisfied diversity even when a smaller number of sampling steps is used. A classifier-free guidance method is adapted for conditional generation. It is also worthwhile to point out that after generative training, our model can be fine-tuned on regression and classification tasks with the state-of-the-art performance, which opens the gate of building all-in-one models in a single module style. Our model has been open sourced at https://github.com/Augus1999/bayesian-flow-network-for-chemistry.

Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.


GemNet-OC: Developing Graph Neural Networks for Large and Diverse Molecular Simulation Datasets
Recent years have seen the advent of molecular simulation datasets that are orders of magnitude larger and more diverse. These new datasets differ substantially in four aspects of complexity: 1. Chemical diversity (number of different elements), 2. system size (number of atoms per sample), 3. dataset size (number of data samples), and 4. domain shift (similarity of the training and test set). Despite these large differences, benchmarks on small and narrow datasets remain the predominant method of demonstrating progress in graph neural networks (GNNs) for molecular simulation, likely due to cheaper training compute requirements. This raises the question -- does GNN progress on small and narrow datasets translate to these more complex datasets? This work investigates this question by first developing the GemNet-OC model based on the large Open Catalyst 2020 (OC20) dataset. GemNet-OC outperforms the previous state-of-the-art on OC20 by 16% while reducing training time by a factor of 10. We then compare the impact of 18 model components and hyperparameter choices on performance in multiple datasets. We find that the resulting model would be drastically different depending on the dataset used for making model choices. To isolate the source of this discrepancy we study six subsets of the OC20 dataset that individually test each of the above-mentioned four dataset aspects. We find that results on the OC-2M subset correlate well with the full OC20 dataset while being substantially cheaper to train on. Our findings challenge the common practice of developing GNNs solely on small datasets, but highlight ways of achieving fast development cycles and generalizable results via moderately-sized, representative datasets such as OC-2M and efficient models such as GemNet-OC. Our code and pretrained model weights are open-sourced.


Matbench Discovery -- An evaluation framework for machine learning crystal stability prediction
Matbench Discovery simulates the deployment of machine learning (ML) energy models in a high-throughput search for stable inorganic crystals. We address the disconnect between (i) thermodynamic stability and formation energy and (ii) in-domain vs out-of-distribution performance. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with further insights into trade-offs between various performance metrics. To answer the question which ML methodology performs best at materials discovery, our initial release explores a variety of models including random forests, graph neural networks (GNN), one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials (UIP). Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find CHGNet > M3GNet > MACE > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi tessellation fingerprints with random forest. The top 3 models are UIPs, the winning methodology for ML-guided materials discovery, achieving F1 scores of ~0.6 for crystal stability classification and discovery acceleration factors (DAF) of up to 5x on the first 10k most stable predictions compared to dummy selection from our test set. We also highlight a sharp disconnect between commonly used global regression metrics and more task-relevant classification metrics. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV/atom above the convex hull where most materials are. Our results highlight the need to focus on classification metrics that actually correlate with improved stability hit rate.
Simplicial Closure and higher-order link prediction
Networks provide a powerful formalism for modeling complex systems by using a model of pairwise interactions. But much of the structure within these systems involves interactions that take place among more than two nodes at once; for example, communication within a group rather than person-to person, collaboration among a team rather than a pair of coauthors, or biological interaction between a set of molecules rather than just two. Such higher-order interactions are ubiquitous, but their empirical study has received limited attention, and little is known about possible organizational principles of such structures. Here we study the temporal evolution of 19 datasets with explicit accounting for higher-order interactions. We show that there is a rich variety of structure in our datasets but datasets from the same system types have consistent patterns of higher-order structure. Furthermore, we find that tie strength and edge density are competing positive indicators of higher-order organization, and these trends are consistent across interactions involving differing numbers of nodes. To systematically further the study of theories for such higher-order structures, we propose higher-order link prediction as a benchmark problem to assess models and algorithms that predict higher-order structure. We find a fundamental differences from traditional pairwise link prediction, with a greater role for local rather than long-range information in predicting the appearance of new interactions.
Beyond the Selected Completely At Random Assumption for Learning from Positive and Unlabeled Data
Most positive and unlabeled data is subject to selection biases. The labeled examples can, for example, be selected from the positive set because they are easier to obtain or more obviously positive. This paper investigates how learning can be ena BHbled in this setting. We propose and theoretically analyze an empirical-risk-based method for incorporating the labeling mechanism. Additionally, we investigate under which assumptions learning is possible when the labeling mechanism is not fully understood and propose a practical method to enable this. Our empirical analysis supports the theoretical results and shows that taking into account the possibility of a selection bias, even when the labeling mechanism is unknown, improves the trained classifiers.
Weighted least-squares approximation with determinantal point processes and generalized volume sampling
We consider the problem of approximating a function from L^2 by an element of a given m-dimensional space V_m, associated with some feature map varphi, using evaluations of the function at random points x_1,dots,x_n. After recalling some results on optimal weighted least-squares using independent and identically distributed points, we consider weighted least-squares using projection determinantal point processes (DPP) or volume sampling. These distributions introduce dependence between the points that promotes diversity in the selected features varphi(x_i). We first provide a generalized version of volume-rescaled sampling yielding quasi-optimality results in expectation with a number of samples n = O(mlog(m)), that means that the expected L^2 error is bounded by a constant times the best approximation error in L^2. Also, further assuming that the function is in some normed vector space H continuously embedded in L^2, we further prove that the approximation is almost surely bounded by the best approximation error measured in the H-norm. This includes the cases of functions from L^infty or reproducing kernel Hilbert spaces. Finally, we present an alternative strategy consisting in using independent repetitions of projection DPP (or volume sampling), yielding similar error bounds as with i.i.d. or volume sampling, but in practice with a much lower number of samples. Numerical experiments illustrate the performance of the different strategies.
Robust Model-Based Optimization for Challenging Fitness Landscapes
Protein design, a grand challenge of the day, involves optimization on a fitness landscape, and leading methods adopt a model-based approach where a model is trained on a training set (protein sequences and fitness) and proposes candidates to explore next. These methods are challenged by sparsity of high-fitness samples in the training set, a problem that has been in the literature. A less recognized but equally important problem stems from the distribution of training samples in the design space: leading methods are not designed for scenarios where the desired optimum is in a region that is not only poorly represented in training data, but also relatively far from the highly represented low-fitness regions. We show that this problem of "separation" in the design space is a significant bottleneck in existing model-based optimization tools and propose a new approach that uses a novel VAE as its search model to overcome the problem. We demonstrate its advantage over prior methods in robustly finding improved samples, regardless of the imbalance and separation between low- and high-fitness training samples. Our comprehensive benchmark on real and semi-synthetic protein datasets as well as solution design for physics-informed neural networks, showcases the generality of our approach in discrete and continuous design spaces. Our implementation is available at https://github.com/sabagh1994/PGVAE.
Learning Inter-Atomic Potentials without Explicit Equivariance
Accurate and scalable machine-learned inter-atomic potentials (MLIPs) are essential for molecular simulations ranging from drug discovery to new material design. Current state-of-the-art models enforce roto-translational symmetries through equivariant neural network architectures, a hard-wired inductive bias that can often lead to reduced flexibility, computational efficiency, and scalability. In this work, we introduce TransIP: Transformer-based Inter-Atomic Potentials, a novel training paradigm for interatomic potentials achieving symmetry compliance without explicit architectural constraints. Our approach guides a generic non-equivariant Transformer-based model to learn SO(3)-equivariance by optimizing its representations in the embedding space. Trained on the recent Open Molecules (OMol25) collection, a large and diverse molecular dataset built specifically for MLIPs and covering different types of molecules (including small organics, biomolecular fragments, and electrolyte-like species), TransIP attains comparable performance in machine-learning force fields versus state-of-the-art equivariant baselines. Further, compared to a data augmentation baseline, TransIP achieves 40% to 60% improvement in performance across varying OMol25 dataset sizes. More broadly, our work shows that learned equivariance can be a powerful and efficient alternative to equivariant or augmentation-based MLIP models.
SΩI: Score-based O-INFORMATION Estimation
The analysis of scientific data and complex multivariate systems requires information quantities that capture relationships among multiple random variables. Recently, new information-theoretic measures have been developed to overcome the shortcomings of classical ones, such as mutual information, that are restricted to considering pairwise interactions. Among them, the concept of information synergy and redundancy is crucial for understanding the high-order dependencies between variables. One of the most prominent and versatile measures based on this concept is O-information, which provides a clear and scalable way to quantify the synergy-redundancy balance in multivariate systems. However, its practical application is limited to simplified cases. In this work, we introduce SOmegaI, which allows for the first time to compute O-information without restrictive assumptions about the system. Our experiments validate our approach on synthetic data, and demonstrate the effectiveness of SOmegaI in the context of a real-world use case.

A Robust Ensemble Algorithm for Ischemic Stroke Lesion Segmentation: Generalizability and Clinical Utility Beyond the ISLES Challenge
Diffusion-weighted MRI (DWI) is essential for stroke diagnosis, treatment decisions, and prognosis. However, image and disease variability hinder the development of generalizable AI algorithms with clinical value. We address this gap by presenting a novel ensemble algorithm derived from the 2022 Ischemic Stroke Lesion Segmentation (ISLES) challenge. ISLES'22 provided 400 patient scans with ischemic stroke from various medical centers, facilitating the development of a wide range of cutting-edge segmentation algorithms by the research community. Through collaboration with leading teams, we combined top-performing algorithms into an ensemble model that overcomes the limitations of individual solutions. Our ensemble model achieved superior ischemic lesion detection and segmentation accuracy on our internal test set compared to individual algorithms. This accuracy generalized well across diverse image and disease variables. Furthermore, the model excelled in extracting clinical biomarkers. Notably, in a Turing-like test, neuroradiologists consistently preferred the algorithm's segmentations over manual expert efforts, highlighting increased comprehensiveness and precision. Validation using a real-world external dataset (N=1686) confirmed the model's generalizability. The algorithm's outputs also demonstrated strong correlations with clinical scores (admission NIHSS and 90-day mRS) on par with or exceeding expert-derived results, underlining its clinical relevance. This study offers two key findings. First, we present an ensemble algorithm (https://github.com/Tabrisrei/ISLES22_Ensemble) that detects and segments ischemic stroke lesions on DWI across diverse scenarios on par with expert (neuro)radiologists. Second, we show the potential for biomedical challenge outputs to extend beyond the challenge's initial objectives, demonstrating their real-world clinical applicability.

DriftMoE: A Mixture of Experts Approach to Handle Concept Drifts
Learning from non-stationary data streams subject to concept drift requires models that can adapt on-the-fly while remaining resource-efficient. Existing adaptive ensemble methods often rely on coarse-grained adaptation mechanisms or simple voting schemes that fail to optimally leverage specialized knowledge. This paper introduces DriftMoE, an online Mixture-of-Experts (MoE) architecture that addresses these limitations through a novel co-training framework. DriftMoE features a compact neural router that is co-trained alongside a pool of incremental Hoeffding tree experts. The key innovation lies in a symbiotic learning loop that enables expert specialization: the router selects the most suitable expert for prediction, the relevant experts update incrementally with the true label, and the router refines its parameters using a multi-hot correctness mask that reinforces every accurate expert. This feedback loop provides the router with a clear training signal while accelerating expert specialization. We evaluate DriftMoE's performance across nine state-of-the-art data stream learning benchmarks spanning abrupt, gradual, and real-world drifts testing two distinct configurations: one where experts specialize on data regimes (multi-class variant), and another where they focus on single-class specialization (task-based variant). Our results demonstrate that DriftMoE achieves competitive results with state-of-the-art stream learning adaptive ensembles, offering a principled and efficient approach to concept drift adaptation. All code, data pipelines, and reproducibility scripts are available in our public GitHub repository: https://github.com/miguel-ceadar/drift-moe.



The Open Catalyst 2020 (OC20) Dataset and Community Challenges
Catalyst discovery and optimization is key to solving many societal and energy challenges including solar fuels synthesis, long-term energy storage, and renewable fertilizer production. Despite considerable effort by the catalysis community to apply machine learning models to the computational catalyst discovery process, it remains an open challenge to build models that can generalize across both elemental compositions of surfaces and adsorbate identity/configurations, perhaps because datasets have been smaller in catalysis than related fields. To address this we developed the OC20 dataset, consisting of 1,281,040 Density Functional Theory (DFT) relaxations (~264,890,000 single point evaluations) across a wide swath of materials, surfaces, and adsorbates (nitrogen, carbon, and oxygen chemistries). We supplemented this dataset with randomly perturbed structures, short timescale molecular dynamics, and electronic structure analyses. The dataset comprises three central tasks indicative of day-to-day catalyst modeling and comes with pre-defined train/validation/test splits to facilitate direct comparisons with future model development efforts. We applied three state-of-the-art graph neural network models (CGCNN, SchNet, Dimenet++) to each of these tasks as baseline demonstrations for the community to build on. In almost every task, no upper limit on model size was identified, suggesting that even larger models are likely to improve on initial results. The dataset and baseline models are both provided as open resources, as well as a public leader board to encourage community contributions to solve these important tasks.
Batch Predictive Inference
Constructing prediction sets with coverage guarantees for unobserved outcomes is a core problem in modern statistics. Methods for predictive inference have been developed for a wide range of settings, but usually only consider test data points one at a time. Here we study the problem of distribution-free predictive inference for a batch of multiple test points, aiming to construct prediction sets for functions -- such as the mean or median -- of any number of unobserved test datapoints. This setting includes constructing simultaneous prediction sets with a high probability of coverage, and selecting datapoints satisfying a specified condition while controlling the number of false claims. For the general task of predictive inference on a function of a batch of test points, we introduce a methodology called batch predictive inference (batch PI), and provide a distribution-free coverage guarantee under exchangeability of the calibration and test data. Batch PI requires the quantiles of a rank ordering function defined on certain subsets of ranks. While computing these quantiles is NP-hard in general, we show that it can be done efficiently in many cases of interest, most notably for batch score functions with a compositional structure -- which includes examples of interest such as the mean -- via a dynamic programming algorithm that we develop. Batch PI has advantages over naive approaches (such as partitioning the calibration data or directly extending conformal prediction) in many settings, as it can deliver informative prediction sets even using small calibration sample sizes. We illustrate that our procedures provide informative inference across the use cases mentioned above, through experiments on both simulated data and a drug-target interaction dataset.

iBitter-Stack: A Multi-Representation Ensemble Learning Model for Accurate Bitter Peptide Identification
The identification of bitter peptides is crucial in various domains, including food science, drug discovery, and biochemical research. These peptides not only contribute to the undesirable taste of hydrolyzed proteins but also play key roles in physiological and pharmacological processes. However, experimental methods for identifying bitter peptides are time-consuming and expensive. With the rapid expansion of peptide sequence databases in the post-genomic era, the demand for efficient computational approaches to distinguish bitter from non-bitter peptides has become increasingly significant. In this study, we propose a novel stacking-based ensemble learning framework aimed at enhancing the accuracy and reliability of bitter peptide classification. Our method integrates diverse sequence-based feature representations and leverages a broad set of machine learning classifiers. The first stacking layer comprises multiple base classifiers, each trained on distinct feature encoding schemes, while the second layer employs logistic regression to refine predictions using an eight-dimensional probability vector. Extensive evaluations on a carefully curated dataset demonstrate that our model significantly outperforms existing predictive methods, providing a robust and reliable computational tool for bitter peptide identification. Our approach achieves an accuracy of 96.09\% and a Matthews Correlation Coefficient (MCC) of 0.9220 on the independent test set, underscoring its effectiveness and generalizability. To facilitate real-time usage and broader accessibility, we have also developed a user-friendly web server based on the proposed method, which is freely accessible at https://ibitter-stack-webserver.streamlit.app/. This tool enables researchers and practitioners to conveniently screen peptide sequences for bitterness in real-time applications.

mdCATH: A Large-Scale MD Dataset for Data-Driven Computational Biophysics
Recent advancements in protein structure determination are revolutionizing our understanding of proteins. Still, a significant gap remains in the availability of comprehensive datasets that focus on the dynamics of proteins, which are crucial for understanding protein function, folding, and interactions. To address this critical gap, we introduce mdCATH, a dataset generated through an extensive set of all-atom molecular dynamics simulations of a diverse and representative collection of protein domains. This dataset comprises all-atom systems for 5,398 domains, modeled with a state-of-the-art classical force field, and simulated in five replicates each at five temperatures from 320 K to 413 K. The mdCATH dataset records coordinates and forces every 1 ns, for over 62 ms of accumulated simulation time, effectively capturing the dynamics of the various classes of domains and providing a unique resource for proteome-wide statistical analyses of protein unfolding thermodynamics and kinetics. We outline the dataset structure and showcase its potential through four easily reproducible case studies, highlighting its capabilities in advancing protein science.
Amortized Sampling with Transferable Normalizing Flows
Efficient equilibrium sampling of molecular conformations remains a core challenge in computational chemistry and statistical inference. Classical approaches such as molecular dynamics or Markov chain Monte Carlo inherently lack amortization; the computational cost of sampling must be paid in-full for each system of interest. The widespread success of generative models has inspired interest into overcoming this limitation through learning sampling algorithms. Despite performing on par with conventional methods when trained on a single system, learned samplers have so far demonstrated limited ability to transfer across systems. We prove that deep learning enables the design of scalable and transferable samplers by introducing Prose, a 280 million parameter all-atom transferable normalizing flow trained on a corpus of peptide molecular dynamics trajectories up to 8 residues in length. Prose draws zero-shot uncorrelated proposal samples for arbitrary peptide systems, achieving the previously intractable transferability across sequence length, whilst retaining the efficient likelihood evaluation of normalizing flows. Through extensive empirical evaluation we demonstrate the efficacy of Prose as a proposal for a variety of sampling algorithms, finding a simple importance sampling-based finetuning procedure to achieve superior performance to established methods such as sequential Monte Carlo on unseen tetrapeptides. We open-source the Prose codebase, model weights, and training dataset, to further stimulate research into amortized sampling methods and finetuning objectives.
Understanding and Mitigating Distribution Shifts For Machine Learning Force Fields
Machine Learning Force Fields (MLFFs) are a promising alternative to expensive ab initio quantum mechanical molecular simulations. Given the diversity of chemical spaces that are of interest and the cost of generating new data, it is important to understand how MLFFs generalize beyond their training distributions. In order to characterize and better understand distribution shifts in MLFFs, we conduct diagnostic experiments on chemical datasets, revealing common shifts that pose significant challenges, even for large foundation models trained on extensive data. Based on these observations, we hypothesize that current supervised training methods inadequately regularize MLFFs, resulting in overfitting and learning poor representations of out-of-distribution systems. We then propose two new methods as initial steps for mitigating distribution shifts for MLFFs. Our methods focus on test-time refinement strategies that incur minimal computational cost and do not use expensive ab initio reference labels. The first strategy, based on spectral graph theory, modifies the edges of test graphs to align with graph structures seen during training. Our second strategy improves representations for out-of-distribution systems at test-time by taking gradient steps using an auxiliary objective, such as a cheap physical prior. Our test-time refinement strategies significantly reduce errors on out-of-distribution systems, suggesting that MLFFs are capable of and can move towards modeling diverse chemical spaces, but are not being effectively trained to do so. Our experiments establish clear benchmarks for evaluating the generalization capabilities of the next generation of MLFFs. Our code is available at https://tkreiman.github.io/projects/mlff_distribution_shifts/.


AQCat25: Unlocking spin-aware, high-fidelity machine learning potentials for heterogeneous catalysis
Large-scale datasets have enabled highly accurate machine learning interatomic potentials (MLIPs) for general-purpose heterogeneous catalysis modeling. There are, however, some limitations in what can be treated with these potentials because of gaps in the underlying training data. To extend these capabilities, we introduce AQCat25, a complementary dataset of 13.5 million density functional theory (DFT) single point calculations designed to improve the treatment of systems where spin polarization and/or higher fidelity are critical. We also investigate methodologies for integrating new datasets, such as AQCat25, with the broader Open Catalyst 2020 (OC20) dataset to create spin-aware models without sacrificing generalizability. We find that directly tuning a general model on AQCat25 leads to catastrophic forgetting of the original dataset's knowledge. Conversely, joint training strategies prove effective for improving accuracy on the new data without sacrificing general performance. This joint approach introduces a challenge, as the model must learn from a dataset containing both mixed-fidelity calculations and mixed-physics (spin-polarized vs. unpolarized). We show that explicitly conditioning the model on this system-specific metadata, for example by using Feature-wise Linear Modulation (FiLM), successfully addresses this challenge and further enhances model accuracy. Ultimately, our work establishes an effective protocol for bridging DFT fidelity domains to advance the predictive power of foundational models in catalysis.
A Configurable Library for Generating and Manipulating Maze Datasets
Understanding how machine learning models respond to distributional shifts is a key research challenge. Mazes serve as an excellent testbed due to varied generation algorithms offering a nuanced platform to simulate both subtle and pronounced distributional shifts. To enable systematic investigations of model behavior on out-of-distribution data, we present maze-dataset, a comprehensive library for generating, processing, and visualizing datasets consisting of maze-solving tasks. With this library, researchers can easily create datasets, having extensive control over the generation algorithm used, the parameters fed to the algorithm of choice, and the filters that generated mazes must satisfy. Furthermore, it supports multiple output formats, including rasterized and text-based, catering to convolutional neural networks and autoregressive transformer models. These formats, along with tools for visualizing and converting between them, ensure versatility and adaptability in research applications.



Is Model Ensemble Necessary? Model-based RL via a Single Model with Lipschitz Regularized Value Function
Probabilistic dynamics model ensemble is widely used in existing model-based reinforcement learning methods as it outperforms a single dynamics model in both asymptotic performance and sample efficiency. In this paper, we provide both practical and theoretical insights on the empirical success of the probabilistic dynamics model ensemble through the lens of Lipschitz continuity. We find that, for a value function, the stronger the Lipschitz condition is, the smaller the gap between the true dynamics- and learned dynamics-induced Bellman operators is, thus enabling the converged value function to be closer to the optimal value function. Hence, we hypothesize that the key functionality of the probabilistic dynamics model ensemble is to regularize the Lipschitz condition of the value function using generated samples. To test this hypothesis, we devise two practical robust training mechanisms through computing the adversarial noise and regularizing the value network's spectral norm to directly regularize the Lipschitz condition of the value functions. Empirical results show that combined with our mechanisms, model-based RL algorithms with a single dynamics model outperform those with an ensemble of probabilistic dynamics models. These findings not only support the theoretical insight, but also provide a practical solution for developing computationally efficient model-based RL algorithms.


In-Dataset Trajectory Return Regularization for Offline Preference-based Reinforcement Learning
Offline preference-based reinforcement learning (PbRL) typically operates in two phases: first, use human preferences to learn a reward model and annotate rewards for a reward-free offline dataset; second, learn a policy by optimizing the learned reward via offline RL. However, accurately modeling step-wise rewards from trajectory-level preference feedback presents inherent challenges. The reward bias introduced, particularly the overestimation of predicted rewards, leads to optimistic trajectory stitching, which undermines the pessimism mechanism critical to the offline RL phase. To address this challenge, we propose In-Dataset Trajectory Return Regularization (DTR) for offline PbRL, which leverages conditional sequence modeling to mitigate the risk of learning inaccurate trajectory stitching under reward bias. Specifically, DTR employs Decision Transformer and TD-Learning to strike a balance between maintaining fidelity to the behavior policy with high in-dataset trajectory returns and selecting optimal actions based on high reward labels. Additionally, we introduce an ensemble normalization technique that effectively integrates multiple reward models, balancing the tradeoff between reward differentiation and accuracy. Empirical evaluations on various benchmarks demonstrate the superiority of DTR over other state-of-the-art baselines.

Deep Learning for Protein-Ligand Docking: Are We There Yet?
The effects of ligand binding on protein structures and their in vivo functions carry numerous implications for modern biomedical research and biotechnology development efforts such as drug discovery. Although several deep learning (DL) methods and benchmarks designed for protein-ligand docking have recently been introduced, to date no prior works have systematically studied the behavior of the latest docking and structure prediction methods within the broadly applicable context of (1) using predicted (apo) protein structures for docking (e.g., for applicability to new proteins); (2) binding multiple (cofactor) ligands concurrently to a given target protein (e.g., for enzyme design); and (3) having no prior knowledge of binding pockets (e.g., for generalization to unknown pockets). To enable a deeper understanding of docking methods' real-world utility, we introduce PoseBench, the first comprehensive benchmark for broadly applicable protein-ligand docking. PoseBench enables researchers to rigorously and systematically evaluate DL methods for apo-to-holo protein-ligand docking and protein-ligand structure prediction using both primary ligand and multi-ligand benchmark datasets, the latter of which we introduce for the first time to the DL community. Empirically, using PoseBench, we find that (1) DL co-folding methods generally outperform comparable conventional and DL docking baselines, yet popular methods such as AlphaFold 3 are still challenged by prediction targets with novel protein sequences; (2) certain DL co-folding methods are highly sensitive to their input multiple sequence alignments, while others are not; and (3) DL methods struggle to strike a balance between structural accuracy and chemical specificity when predicting novel or multi-ligand protein targets. Code, data, tutorials, and benchmark results are available at https://github.com/BioinfoMachineLearning/PoseBench.

Repelling Random Walks
We present a novel quasi-Monte Carlo mechanism to improve graph-based sampling, coined repelling random walks. By inducing correlations between the trajectories of an interacting ensemble such that their marginal transition probabilities are unmodified, we are able to explore the graph more efficiently, improving the concentration of statistical estimators whilst leaving them unbiased. The mechanism has a trivial drop-in implementation. We showcase the effectiveness of repelling random walks in a range of settings including estimation of graph kernels, the PageRank vector and graphlet concentrations. We provide detailed experimental evaluation and robust theoretical guarantees. To our knowledge, repelling random walks constitute the first rigorously studied quasi-Monte Carlo scheme correlating the directions of walkers on a graph, inviting new research in this exciting nascent domain.
MeLM, a generative pretrained language modeling framework that solves forward and inverse mechanics problems
We report a flexible multi-modal mechanics language model, MeLM, applied to solve various nonlinear forward and inverse problems, that can deal with a set of instructions, numbers and microstructure data. The framework is applied to various examples including bio-inspired hierarchical honeycomb design, carbon nanotube mechanics, and protein unfolding. In spite of the flexible nature of the model-which allows us to easily incorporate diverse materials, scales, and mechanical features-it performs well across disparate forward and inverse tasks. Based on an autoregressive attention-model, MeLM effectively represents a large multi-particle system consisting of hundreds of millions of neurons, where the interaction potentials are discovered through graph-forming self-attention mechanisms that are then used to identify relationships from emergent structures, while taking advantage of synergies discovered in the training data. We show that the model can solve complex degenerate mechanics design problems and determine novel material architectures across a range of hierarchical levels, providing an avenue for materials discovery and analysis. Looking beyond the demonstrations reported in this paper, we discuss other opportunities in applied mechanics and general considerations about the use of large language models in modeling, design, and analysis that can span a broad spectrum of material properties from mechanical, thermal, optical, to electronic.
Graphically Structured Diffusion Models
We introduce a framework for automatically defining and learning deep generative models with problem-specific structure. We tackle problem domains that are more traditionally solved by algorithms such as sorting, constraint satisfaction for Sudoku, and matrix factorization. Concretely, we train diffusion models with an architecture tailored to the problem specification. This problem specification should contain a graphical model describing relationships between variables, and often benefits from explicit representation of subcomputations. Permutation invariances can also be exploited. Across a diverse set of experiments we improve the scaling relationship between problem dimension and our model's performance, in terms of both training time and final accuracy. Our code can be found at https://github.com/plai-group/gsdm.

ProteinBench: A Holistic Evaluation of Protein Foundation Models
Recent years have witnessed a surge in the development of protein foundation models, significantly improving performance in protein prediction and generative tasks ranging from 3D structure prediction and protein design to conformational dynamics. However, the capabilities and limitations associated with these models remain poorly understood due to the absence of a unified evaluation framework. To fill this gap, we introduce ProteinBench, a holistic evaluation framework designed to enhance the transparency of protein foundation models. Our approach consists of three key components: (i) A taxonomic classification of tasks that broadly encompass the main challenges in the protein domain, based on the relationships between different protein modalities; (ii) A multi-metric evaluation approach that assesses performance across four key dimensions: quality, novelty, diversity, and robustness; and (iii) In-depth analyses from various user objectives, providing a holistic view of model performance. Our comprehensive evaluation of protein foundation models reveals several key findings that shed light on their current capabilities and limitations. To promote transparency and facilitate further research, we release the evaluation dataset, code, and a public leaderboard publicly for further analysis and a general modular toolkit. We intend for ProteinBench to be a living benchmark for establishing a standardized, in-depth evaluation framework for protein foundation models, driving their development and application while fostering collaboration within the field.




The Power of Few: Accelerating and Enhancing Data Reweighting with Coreset Selection
As machine learning tasks continue to evolve, the trend has been to gather larger datasets and train increasingly larger models. While this has led to advancements in accuracy, it has also escalated computational costs to unsustainable levels. Addressing this, our work aims to strike a delicate balance between computational efficiency and model accuracy, a persisting challenge in the field. We introduce a novel method that employs core subset selection for reweighting, effectively optimizing both computational time and model performance. By focusing on a strategically selected coreset, our approach offers a robust representation, as it efficiently minimizes the influence of outliers. The re-calibrated weights are then mapped back to and propagated across the entire dataset. Our experimental results substantiate the effectiveness of this approach, underscoring its potential as a scalable and precise solution for model training.

Towards Best Practices of Activation Patching in Language Models: Metrics and Methods
Mechanistic interpretability seeks to understand the internal mechanisms of machine learning models, where localization -- identifying the important model components -- is a key step. Activation patching, also known as causal tracing or interchange intervention, is a standard technique for this task (Vig et al., 2020), but the literature contains many variants with little consensus on the choice of hyperparameters or methodology. In this work, we systematically examine the impact of methodological details in activation patching, including evaluation metrics and corruption methods. In several settings of localization and circuit discovery in language models, we find that varying these hyperparameters could lead to disparate interpretability results. Backed by empirical observations, we give conceptual arguments for why certain metrics or methods may be preferred. Finally, we provide recommendations for the best practices of activation patching going forwards.


Algorithmic Collective Action in Machine Learning
We initiate a principled study of algorithmic collective action on digital platforms that deploy machine learning algorithms. We propose a simple theoretical model of a collective interacting with a firm's learning algorithm. The collective pools the data of participating individuals and executes an algorithmic strategy by instructing participants how to modify their own data to achieve a collective goal. We investigate the consequences of this model in three fundamental learning-theoretic settings: the case of a nonparametric optimal learning algorithm, a parametric risk minimizer, and gradient-based optimization. In each setting, we come up with coordinated algorithmic strategies and characterize natural success criteria as a function of the collective's size. Complementing our theory, we conduct systematic experiments on a skill classification task involving tens of thousands of resumes from a gig platform for freelancers. Through more than two thousand model training runs of a BERT-like language model, we see a striking correspondence emerge between our empirical observations and the predictions made by our theory. Taken together, our theory and experiments broadly support the conclusion that algorithmic collectives of exceedingly small fractional size can exert significant control over a platform's learning algorithm.
Accuracy on the Curve: On the Nonlinear Correlation of ML Performance Between Data Subpopulations
Understanding the performance of machine learning (ML) models across diverse data distributions is critically important for reliable applications. Despite recent empirical studies positing a near-perfect linear correlation between in-distribution (ID) and out-of-distribution (OOD) accuracies, we empirically demonstrate that this correlation is more nuanced under subpopulation shifts. Through rigorous experimentation and analysis across a variety of datasets, models, and training epochs, we demonstrate that OOD performance often has a nonlinear correlation with ID performance in subpopulation shifts. Our findings, which contrast previous studies that have posited a linear correlation in model performance during distribution shifts, reveal a "moon shape" correlation (parabolic uptrend curve) between the test performance on the majority subpopulation and the minority subpopulation. This non-trivial nonlinear correlation holds across model architectures, hyperparameters, training durations, and the imbalance between subpopulations. Furthermore, we found that the nonlinearity of this "moon shape" is causally influenced by the degree of spurious correlations in the training data. Our controlled experiments show that stronger spurious correlation in the training data creates more nonlinear performance correlation. We provide complementary experimental and theoretical analyses for this phenomenon, and discuss its implications for ML reliability and fairness. Our work highlights the importance of understanding the nonlinear effects of model improvement on performance in different subpopulations, and has the potential to inform the development of more equitable and responsible machine learning models.

X-LoRA: Mixture of Low-Rank Adapter Experts, a Flexible Framework for Large Language Models with Applications in Protein Mechanics and Design
We report a mixture of expert strategy to create fine-tuned large language models using a deep layer-wise token-level approach based on low-rank adaptation (LoRA). Starting with a set of pre-trained LoRA adapters, we propose a gating strategy that uses the hidden states to dynamically mix adapted layers, allowing the resulting X-LoRA model to draw upon different capabilities and create never-before-used deep layer-wise combinations of adaptations are established to solve specific tasks. The design is inspired by the biological principles of universality and diversity, where neural network building blocks are reused in different hierarchical manifestations. Hence, the X-LoRA model can be easily implemented for any existing large language model (LLM) without a need for modifications of the underlying structure. We develop a tailored X-LoRA model that offers scientific capabilities including forward/inverse analysis tasks and enhanced reasoning capability, focused on biomaterial analysis, protein mechanics and design. The impact of this work include access to readily expandable, adaptable and changeable models with strong domain knowledge and the capability to integrate across areas of knowledge. With the X-LoRA model featuring experts in biology, mathematics, reasoning, bio-inspired materials, mechanics and materials, chemistry, and protein mechanics we conduct a series of physics-focused case studies. We examine knowledge recall, protein mechanics forward/inverse tasks, protein design, and adversarial agentic modeling including ontological knowledge graphs. The model is capable not only of making quantitative predictions of nanomechanical properties of proteins, but also reasons over the results and correctly predicts likely mechanisms that explain distinct molecular behaviors.
In-Context Learning Strategies Emerge Rationally
Recent work analyzing in-context learning (ICL) has identified a broad set of strategies that describe model behavior in different experimental conditions. We aim to unify these findings by asking why a model learns these disparate strategies in the first place. Specifically, we start with the observation that when trained to learn a mixture of tasks, as is popular in the literature, the strategies learned by a model for performing ICL can be captured by a family of Bayesian predictors: a memorizing predictor, which assumes a discrete prior on the set of seen tasks, and a generalizing predictor, where the prior matches the underlying task distribution. Adopting the normative lens of rational analysis, where a learner's behavior is explained as an optimal adaptation to data given computational constraints, we develop a hierarchical Bayesian framework that almost perfectly predicts Transformer next-token predictions throughout training -- without assuming access to its weights. Under this framework, pretraining is viewed as a process of updating the posterior probability of different strategies, and inference-time behavior as a posterior-weighted average over these strategies' predictions. Our framework draws on common assumptions about neural network learning dynamics, which make explicit a tradeoff between loss and complexity among candidate strategies: beyond how well it explains the data, a model's preference towards implementing a strategy is dictated by its complexity. This helps explain well-known ICL phenomena, while offering novel predictions: e.g., we show a superlinear trend in the timescale for transitioning from generalization to memorization as task diversity increases. Overall, our work advances an explanatory and predictive account of ICL grounded in tradeoffs between strategy loss and complexity.

Returning The Favour: When Regression Benefits From Probabilistic Causal Knowledge
A directed acyclic graph (DAG) provides valuable prior knowledge that is often discarded in regression tasks in machine learning. We show that the independences arising from the presence of collider structures in DAGs provide meaningful inductive biases, which constrain the regression hypothesis space and improve predictive performance. We introduce collider regression, a framework to incorporate probabilistic causal knowledge from a collider in a regression problem. When the hypothesis space is a reproducing kernel Hilbert space, we prove a strictly positive generalisation benefit under mild assumptions and provide closed-form estimators of the empirical risk minimiser. Experiments on synthetic and climate model data demonstrate performance gains of the proposed methodology.
Symmetry-invariant quantum machine learning force fields
Machine learning techniques are essential tools to compute efficient, yet accurate, force fields for atomistic simulations. This approach has recently been extended to incorporate quantum computational methods, making use of variational quantum learning models to predict potential energy surfaces and atomic forces from ab initio training data. However, the trainability and scalability of such models are still limited, due to both theoretical and practical barriers. Inspired by recent developments in geometric classical and quantum machine learning, here we design quantum neural networks that explicitly incorporate, as a data-inspired prior, an extensive set of physically relevant symmetries. We find that our invariant quantum learning models outperform their more generic counterparts on individual molecules of growing complexity. Furthermore, we study a water dimer as a minimal example of a system with multiple components, showcasing the versatility of our proposed approach and opening the way towards larger simulations. Our results suggest that molecular force fields generation can significantly profit from leveraging the framework of geometric quantum machine learning, and that chemical systems represent, in fact, an interesting and rich playground for the development and application of advanced quantum machine learning tools.
Pep2Prob Benchmark: Predicting Fragment Ion Probability for MS^2-based Proteomics
Proteins perform nearly all cellular functions and constitute most drug targets, making their analysis fundamental to understanding human biology in health and disease. Tandem mass spectrometry (MS^2) is the major analytical technique in proteomics that identifies peptides by ionizing them, fragmenting them, and using the resulting mass spectra to identify and quantify proteins in biological samples. In MS^2 analysis, peptide fragment ion probability prediction plays a critical role, enhancing the accuracy of peptide identification from mass spectra as a complement to the intensity information. Current approaches rely on global statistics of fragmentation, which assumes that a fragment's probability is uniform across all peptides. Nevertheless, this assumption is oversimplified from a biochemical principle point of view and limits accurate prediction. To address this gap, we present Pep2Prob, the first comprehensive dataset and benchmark designed for peptide-specific fragment ion probability prediction. The proposed dataset contains fragment ion probability statistics for 608,780 unique precursors (each precursor is a pair of peptide sequence and charge state), summarized from more than 183 million high-quality, high-resolution, HCD MS^2 spectra with validated peptide assignments and fragmentation annotations. We establish baseline performance using simple statistical rules and learning-based methods, and find that models leveraging peptide-specific information significantly outperform previous methods using only global fragmentation statistics. Furthermore, performance across benchmark models with increasing capacities suggests that the peptide-fragmentation relationship exhibits complex nonlinearities requiring sophisticated machine learning approaches.
Rethinking the "Heatmap + Monte Carlo Tree Search" Paradigm for Solving Large Scale TSP
The Travelling Salesman Problem (TSP) remains a fundamental challenge in combinatorial optimization, inspiring diverse algorithmic strategies. This paper revisits the "heatmap + Monte Carlo Tree Search (MCTS)" paradigm that has recently gained traction for learning-based TSP solutions. Within this framework, heatmaps encode the likelihood of edges forming part of the optimal tour, and MCTS refines this probabilistic guidance to discover optimal solutions. Contemporary approaches have predominantly emphasized the refinement of heatmap generation through sophisticated learning models, inadvertently sidelining the critical role of MCTS. Our extensive empirical analysis reveals two pivotal insights: 1) The configuration of MCTS strategies profoundly influences the solution quality, demanding meticulous tuning to leverage their full potential; 2) Our findings demonstrate that a rudimentary and parameter-free heatmap, derived from the intrinsic k-nearest nature of TSP, can rival or even surpass the performance of complicated heatmaps, with strong generalizability across various scales. Empirical evaluations across various TSP scales underscore the efficacy of our approach, achieving competitive results. These observations challenge the prevailing focus on heatmap sophistication, advocating a reevaluation of the paradigm to harness both components synergistically. Our code is available at: https://github.com/LOGO-CUHKSZ/rethink_mcts_tsp.
MOMENT: A Family of Open Time-series Foundation Models
We introduce MOMENT, a family of open-source foundation models for general-purpose time-series analysis. Pre-training large models on time-series data is challenging due to (1) the absence of a large and cohesive public time-series repository, and (2) diverse time-series characteristics which make multi-dataset training onerous. Additionally, (3) experimental benchmarks to evaluate these models, especially in scenarios with limited resources, time, and supervision, are still in their nascent stages. To address these challenges, we compile a large and diverse collection of public time-series, called the Time-series Pile, and systematically tackle time-series-specific challenges to unlock large-scale multi-dataset pre-training. Finally, we build on recent work to design a benchmark to evaluate time-series foundation models on diverse tasks and datasets in limited supervision settings. Experiments on this benchmark demonstrate the effectiveness of our pre-trained models with minimal data and task-specific fine-tuning. Finally, we present several interesting empirical observations about large pre-trained time-series models. Our code is available anonymously at anonymous.4open.science/r/BETT-773F/.

Less is More: Efficient Black-box Attribution via Minimal Interpretable Subset Selection
To develop a trustworthy AI system, which aim to identify the input regions that most influence the models decisions. The primary task of existing attribution methods lies in efficiently and accurately identifying the relationships among input-prediction interactions. Particularly when the input data is discrete, such as images, analyzing the relationship between inputs and outputs poses a significant challenge due to the combinatorial explosion. In this paper, we propose a novel and efficient black-box attribution mechanism, LiMA (Less input is More faithful for Attribution), which reformulates the attribution of important regions as an optimization problem for submodular subset selection. First, to accurately assess interactions, we design a submodular function that quantifies subset importance and effectively captures their impact on decision outcomes. Then, efficiently ranking input sub-regions by their importance for attribution, we improve optimization efficiency through a novel bidirectional greedy search algorithm. LiMA identifies both the most and least important samples while ensuring an optimal attribution boundary that minimizes errors. Extensive experiments on eight foundation models demonstrate that our method provides faithful interpretations with fewer regions and exhibits strong generalization, shows an average improvement of 36.3% in Insertion and 39.6% in Deletion. Our method also outperforms the naive greedy search in attribution efficiency, being 1.6 times faster. Furthermore, when explaining the reasons behind model prediction errors, the average highest confidence achieved by our method is, on average, 86.1% higher than that of state-of-the-art attribution algorithms. The code is available at https://github.com/RuoyuChen10/LIMA.

BAMBOO: a predictive and transferable machine learning force field framework for liquid electrolyte development
Despite the widespread applications of machine learning force field (MLFF) on solids and small molecules, there is a notable gap in applying MLFF to complex liquid electrolytes. In this work, we introduce BAMBOO (ByteDance AI Molecular Simulation Booster), a novel framework for molecular dynamics (MD) simulations, with a demonstration of its capabilities in the context of liquid electrolytes for lithium batteries. We design a physics-inspired graph equivariant transformer architecture as the backbone of BAMBOO to learn from quantum mechanical simulations. Additionally, we pioneer an ensemble knowledge distillation approach and apply it on MLFFs to improve the stability of MD simulations. Finally, we propose the density alignment algorithm to align BAMBOO with experimental measurements. BAMBOO demonstrates state-of-the-art accuracy in predicting key electrolyte properties such as density, viscosity, and ionic conductivity across various solvents and salt combinations. Our current model, trained on more than 15 chemical species, achieves the average density error of 0.01 g/cm^3 on various compositions compared with experimental data. Moreover, our model demonstrates transferability to molecules not included in the quantum mechanical dataset. We envision this work as paving the way to a "universal MLFF" capable of simulating properties of common organic liquids.
Generative Marginalization Models
We introduce marginalization models (MaMs), a new family of generative models for high-dimensional discrete data. They offer scalable and flexible generative modeling with tractable likelihoods by explicitly modeling all induced marginal distributions. Marginalization models enable fast evaluation of arbitrary marginal probabilities with a single forward pass of the neural network, which overcomes a major limitation of methods with exact marginal inference, such as autoregressive models (ARMs). We propose scalable methods for learning the marginals, grounded in the concept of "marginalization self-consistency". Unlike previous methods, MaMs support scalable training of any-order generative models for high-dimensional problems under the setting of energy-based training, where the goal is to match the learned distribution to a given desired probability (specified by an unnormalized (log) probability function such as energy function or reward function). We demonstrate the effectiveness of the proposed model on a variety of discrete data distributions, including binary images, language, physical systems, and molecules, for maximum likelihood and energy-based training settings. MaMs achieve orders of magnitude speedup in evaluating the marginal probabilities on both settings. For energy-based training tasks, MaMs enable any-order generative modeling of high-dimensional problems beyond the capability of previous methods. Code is at https://github.com/PrincetonLIPS/MaM.
PERFT: Parameter-Efficient Routed Fine-Tuning for Mixture-of-Expert Model
The Mixture-of-Experts (MoE) paradigm has emerged as a powerful approach for scaling transformers with improved resource utilization. However, efficiently fine-tuning MoE models remains largely underexplored. Inspired by recent works on Parameter-Efficient Fine-Tuning (PEFT), we present a unified framework for integrating PEFT modules directly into the MoE mechanism. Aligning with the core principles and architecture of MoE, our framework encompasses a set of design dimensions including various functional and composition strategies. By combining design choices within our framework, we introduce Parameter-Efficient Routed Fine-Tuning (PERFT) as a flexible and scalable family of PEFT strategies tailored for MoE models. Extensive experiments on adapting OLMoE-1B-7B and Mixtral-8times7B for commonsense and arithmetic reasoning tasks demonstrate the effectiveness, scalability, and intriguing dynamics of PERFT. Additionally, we provide empirical findings for each specific design choice to facilitate better application of MoE and PEFT.


IntFold: A Controllable Foundation Model for General and Specialized Biomolecular Structure Prediction
We introduce IntFold, a controllable foundation model for both general and specialized biomolecular structure prediction. IntFold demonstrates predictive accuracy comparable to the state-of-the-art AlphaFold3, while utilizing a superior customized attention kernel. Beyond standard structure prediction, IntFold can be adapted to predict allosteric states, constrained structures, and binding affinity through the use of individual adapters. Furthermore, we introduce a novel confidence head to estimate docking quality, offering a more nuanced assessment for challenging targets such as antibody-antigen complexes. Finally, we share insights gained during the training process of this computationally intensive model.

Leveraging Hyperbolic Embeddings for Coarse-to-Fine Robot Design
Multi-cellular robot design aims to create robots comprised of numerous cells that can be efficiently controlled to perform diverse tasks. Previous research has demonstrated the ability to generate robots for various tasks, but these approaches often optimize robots directly in the vast design space, resulting in robots with complicated morphologies that are hard to control. In response, this paper presents a novel coarse-to-fine method for designing multi-cellular robots. Initially, this strategy seeks optimal coarse-grained robots and progressively refines them. To mitigate the challenge of determining the precise refinement juncture during the coarse-to-fine transition, we introduce the Hyperbolic Embeddings for Robot Design (HERD) framework. HERD unifies robots of various granularity within a shared hyperbolic space and leverages a refined Cross-Entropy Method for optimization. This framework enables our method to autonomously identify areas of exploration in hyperbolic space and concentrate on regions demonstrating promise. Finally, the extensive empirical studies on various challenging tasks sourced from EvoGym show our approach's superior efficiency and generalization capability.

Using Degeneracy in the Loss Landscape for Mechanistic Interpretability
Mechanistic Interpretability aims to reverse engineer the algorithms implemented by neural networks by studying their weights and activations. An obstacle to reverse engineering neural networks is that many of the parameters inside a network are not involved in the computation being implemented by the network. These degenerate parameters may obfuscate internal structure. Singular learning theory teaches us that neural network parameterizations are biased towards being more degenerate, and parameterizations with more degeneracy are likely to generalize further. We identify 3 ways that network parameters can be degenerate: linear dependence between activations in a layer; linear dependence between gradients passed back to a layer; ReLUs which fire on the same subset of datapoints. We also present a heuristic argument that modular networks are likely to be more degenerate, and we develop a metric for identifying modules in a network that is based on this argument. We propose that if we can represent a neural network in a way that is invariant to reparameterizations that exploit the degeneracies, then this representation is likely to be more interpretable, and we provide some evidence that such a representation is likely to have sparser interactions. We introduce the Interaction Basis, a tractable technique to obtain a representation that is invariant to degeneracies from linear dependence of activations or Jacobians.

Do Deep Neural Network Solutions Form a Star Domain?
It has recently been conjectured that neural network solution sets reachable via stochastic gradient descent (SGD) are convex, considering permutation invariances (Entezari et al., 2022). This means that a linear path can connect two independent solutions with low loss, given the weights of one of the models are appropriately permuted. However, current methods to test this theory often require very wide networks to succeed. In this work, we conjecture that more generally, the SGD solution set is a "star domain" that contains a "star model" that is linearly connected to all the other solutions via paths with low loss values, modulo permutations. We propose the Starlight algorithm that finds a star model of a given learning task. We validate our claim by showing that this star model is linearly connected with other independently found solutions. As an additional benefit of our study, we demonstrate better uncertainty estimates on the Bayesian Model Averaging over the obtained star domain. Further, we demonstrate star models as potential substitutes for model ensembles. Our code is available at https://github.com/aktsonthalia/starlight.

Orb-v3: atomistic simulation at scale
We introduce Orb-v3, the next generation of the Orb family of universal interatomic potentials. Models in this family expand the performance-speed-memory Pareto frontier, offering near SoTA performance across a range of evaluations with a >10x reduction in latency and > 8x reduction in memory. Our experiments systematically traverse this frontier, charting the trade-off induced by roto-equivariance, conservatism and graph sparsity. Contrary to recent literature, we find that non-equivariant, non-conservative architectures can accurately model physical properties, including those which require higher-order derivatives of the potential energy surface. This model release is guided by the principle that the most valuable foundation models for atomic simulation will excel on all fronts: accuracy, latency and system size scalability. The reward for doing so is a new era of computational chemistry driven by high-throughput and mesoscale all-atom simulations.
Enhanced Sampling, Public Dataset and Generative Model for Drug-Protein Dissociation Dynamics
Drug-protein binding and dissociation dynamics are fundamental to understanding molecular interactions in biological systems. While many tools for drug-protein interaction studies have emerged, especially artificial intelligence (AI)-based generative models, predictive tools on binding/dissociation kinetics and dynamics are still limited. We propose a novel research paradigm that combines molecular dynamics (MD) simulations, enhanced sampling, and AI generative models to address this issue. We propose an enhanced sampling strategy to efficiently implement the drug-protein dissociation process in MD simulations and estimate the free energy surface (FES). We constructed a program pipeline of MD simulations based on this sampling strategy, thus generating a dataset including 26,612 drug-protein dissociation trajectories containing about 13 million frames. We named this dissociation dynamics dataset DD-13M and used it to train a deep equivariant generative model UnbindingFlow, which can generate collision-free dissociation trajectories. The DD-13M database and UnbindingFlow model represent a significant advancement in computational structural biology, and we anticipate its broad applicability in machine learning studies of drug-protein interactions. Our ongoing efforts focus on expanding this methodology to encompass a broader spectrum of drug-protein complexes and exploring novel applications in pathway prediction.
Dynamic Gaussian Mixture based Deep Generative Model For Robust Forecasting on Sparse Multivariate Time Series
Forecasting on sparse multivariate time series (MTS) aims to model the predictors of future values of time series given their incomplete past, which is important for many emerging applications. However, most existing methods process MTS's individually, and do not leverage the dynamic distributions underlying the MTS's, leading to sub-optimal results when the sparsity is high. To address this challenge, we propose a novel generative model, which tracks the transition of latent clusters, instead of isolated feature representations, to achieve robust modeling. It is characterized by a newly designed dynamic Gaussian mixture distribution, which captures the dynamics of clustering structures, and is used for emitting timeseries. The generative model is parameterized by neural networks. A structured inference network is also designed for enabling inductive analysis. A gating mechanism is further introduced to dynamically tune the Gaussian mixture distributions. Extensive experimental results on a variety of real-life datasets demonstrate the effectiveness of our method.


Interpretable Meta-Learning of Physical Systems
Machine learning methods can be a valuable aid in the scientific process, but they need to face challenging settings where data come from inhomogeneous experimental conditions. Recent meta-learning methods have made significant progress in multi-task learning, but they rely on black-box neural networks, resulting in high computational costs and limited interpretability. Leveraging the structure of the learning problem, we argue that multi-environment generalization can be achieved using a simpler learning model, with an affine structure with respect to the learning task. Crucially, we prove that this architecture can identify the physical parameters of the system, enabling interpreable learning. We demonstrate the competitive generalization performance and the low computational cost of our method by comparing it to state-of-the-art algorithms on physical systems, ranging from toy models to complex, non-analytical systems. The interpretability of our method is illustrated with original applications to physical-parameter-induced adaptation and to adaptive control.
Enhancing Activity Prediction Models in Drug Discovery with the Ability to Understand Human Language
Activity and property prediction models are the central workhorses in drug discovery and materials sciences, but currently they have to be trained or fine-tuned for new tasks. Without training or fine-tuning, scientific language models could be used for such low-data tasks through their announced zero- and few-shot capabilities. However, their predictive quality at activity prediction is lacking. In this work, we envision a novel type of activity prediction model that is able to adapt to new prediction tasks at inference time, via understanding textual information describing the task. To this end, we propose a new architecture with separate modules for chemical and natural language inputs, and a contrastive pre-training objective on data from large biochemical databases. In extensive experiments, we show that our method CLAMP yields improved predictive performance on few-shot learning benchmarks and zero-shot problems in drug discovery. We attribute the advances of our method to the modularized architecture and to our pre-training objective.
Gene-Metabolite Association Prediction with Interactive Knowledge Transfer Enhanced Graph for Metabolite Production
In the rapidly evolving field of metabolic engineering, the quest for efficient and precise gene target identification for metabolite production enhancement presents significant challenges. Traditional approaches, whether knowledge-based or model-based, are notably time-consuming and labor-intensive, due to the vast scale of research literature and the approximation nature of genome-scale metabolic model (GEM) simulations. Therefore, we propose a new task, Gene-Metabolite Association Prediction based on metabolic graphs, to automate the process of candidate gene discovery for a given pair of metabolite and candidate-associated genes, as well as presenting the first benchmark containing 2474 metabolites and 1947 genes of two commonly used microorganisms Saccharomyces cerevisiae (SC) and Issatchenkia orientalis (IO). This task is challenging due to the incompleteness of the metabolic graphs and the heterogeneity among distinct metabolisms. To overcome these limitations, we propose an Interactive Knowledge Transfer mechanism based on Metabolism Graph (IKT4Meta), which improves the association prediction accuracy by integrating the knowledge from different metabolism graphs. First, to build a bridge between two graphs for knowledge transfer, we utilize Pretrained Language Models (PLMs) with external knowledge of genes and metabolites to help generate inter-graph links, significantly alleviating the impact of heterogeneity. Second, we propagate intra-graph links from different metabolic graphs using inter-graph links as anchors. Finally, we conduct the gene-metabolite association prediction based on the enriched metabolism graphs, which integrate the knowledge from multiple microorganisms. Experiments on both types of organisms demonstrate that our proposed methodology outperforms baselines by up to 12.3% across various link prediction frameworks.

Subsample Ridge Ensembles: Equivalences and Generalized Cross-Validation
We study subsampling-based ridge ensembles in the proportional asymptotics regime, where the feature size grows proportionally with the sample size such that their ratio converges to a constant. By analyzing the squared prediction risk of ridge ensembles as a function of the explicit penalty lambda and the limiting subsample aspect ratio phi_s (the ratio of the feature size to the subsample size), we characterize contours in the (lambda, phi_s)-plane at any achievable risk. As a consequence, we prove that the risk of the optimal full ridgeless ensemble (fitted on all possible subsamples) matches that of the optimal ridge predictor. In addition, we prove strong uniform consistency of generalized cross-validation (GCV) over the subsample sizes for estimating the prediction risk of ridge ensembles. This allows for GCV-based tuning of full ridgeless ensembles without sample splitting and yields a predictor whose risk matches optimal ridge risk.

Linear Mode Connectivity in Differentiable Tree Ensembles
Linear Mode Connectivity (LMC) refers to the phenomenon that performance remains consistent for linearly interpolated models in the parameter space. For independently optimized model pairs from different random initializations, achieving LMC is considered crucial for validating the stable success of the non-convex optimization in modern machine learning models and for facilitating practical parameter-based operations such as model merging. While LMC has been achieved for neural networks by considering the permutation invariance of neurons in each hidden layer, its attainment for other models remains an open question. In this paper, we first achieve LMC for soft tree ensembles, which are tree-based differentiable models extensively used in practice. We show the necessity of incorporating two invariances: subtree flip invariance and splitting order invariance, which do not exist in neural networks but are inherent to tree architectures, in addition to permutation invariance of trees. Moreover, we demonstrate that it is even possible to exclude such additional invariances while keeping LMC by designing decision list-based tree architectures, where such invariances do not exist by definition. Our findings indicate the significance of accounting for architecture-specific invariances in achieving LMC.
Deep learning probability flows and entropy production rates in active matter
Active matter systems, from self-propelled colloids to motile bacteria, are characterized by the conversion of free energy into useful work at the microscopic scale. These systems generically involve physics beyond the reach of equilibrium statistical mechanics, and a persistent challenge has been to understand the nature of their nonequilibrium states. The entropy production rate and the magnitude of the steady-state probability current provide quantitative ways to do so by measuring the breakdown of time-reversal symmetry and the strength of nonequilibrium transport of measure. Yet, their efficient computation has remained elusive, as they depend on the system's unknown and high-dimensional probability density. Here, building upon recent advances in generative modeling, we develop a deep learning framework that estimates the score of this density. We show that the score, together with the microscopic equations of motion, gives direct access to the entropy production rate, the probability current, and their decomposition into local contributions from individual particles, spatial regions, and degrees of freedom. To represent the score, we introduce a novel, spatially-local transformer-based network architecture that learns high-order interactions between particles while respecting their underlying permutation symmetry. We demonstrate the broad utility and scalability of the method by applying it to several high-dimensional systems of interacting active particles undergoing motility-induced phase separation (MIPS). We show that a single instance of our network trained on a system of 4096 particles at one packing fraction can generalize to other regions of the phase diagram, including systems with as many as 32768 particles. We use this observation to quantify the spatial structure of the departure from equilibrium in MIPS as a function of the number of particles and the packing fraction.

A Unified Continual Learning Framework with General Parameter-Efficient Tuning
The "pre-training rightarrow downstream adaptation" presents both new opportunities and challenges for Continual Learning (CL). Although the recent state-of-the-art in CL is achieved through Parameter-Efficient-Tuning (PET) adaptation paradigm, only prompt has been explored, limiting its application to Transformers only. In this paper, we position prompting as one instantiation of PET, and propose a unified CL framework with general PET, dubbed as Learning-Accumulation-Ensemble (LAE). PET, e.g., using Adapter, LoRA, or Prefix, can adapt a pre-trained model to downstream tasks with fewer parameters and resources. Given a PET method, our LAE framework incorporates it for CL with three novel designs. 1) Learning: the pre-trained model adapts to the new task by tuning an online PET module, along with our adaptation speed calibration to align different PET modules, 2) Accumulation: the task-specific knowledge learned by the online PET module is accumulated into an offline PET module through momentum update, 3) Ensemble: During inference, we respectively construct two experts with online/offline PET modules (which are favored by the novel/historical tasks) for prediction ensemble. We show that LAE is compatible with a battery of PET methods and gains strong CL capability. For example, LAE with Adaptor PET surpasses the prior state-of-the-art by 1.3% and 3.6% in last-incremental accuracy on CIFAR100 and ImageNet-R datasets, respectively. Code is available at https://github.com/gqk/LAE.
A foundation model for atomistic materials chemistry
Machine-learned force fields have transformed the atomistic modelling of materials by enabling simulations of ab initio quality on unprecedented time and length scales. However, they are currently limited by: (i) the significant computational and human effort that must go into development and validation of potentials for each particular system of interest; and (ii) a general lack of transferability from one chemical system to the next. Here, using the state-of-the-art MACE architecture we introduce a single general-purpose ML model, trained on a public database of 150k inorganic crystals, that is capable of running stable molecular dynamics on molecules and materials. We demonstrate the power of the MACE-MP-0 model -- and its qualitative and at times quantitative accuracy -- on a diverse set problems in the physical sciences, including the properties of solids, liquids, gases, and chemical reactions. The model can be applied out of the box and as a starting or "foundation model" for any atomistic system of interest and is thus a step towards democratising the revolution of ML force fields by lowering the barriers to entry.


Bayesian Flow Is All You Need to Sample Out-of-Distribution Chemical Spaces
Generating novel molecules with higher properties than the training space, namely the out-of-distribution generation, is important for {de~novo} drug design. However, it is not easy for distribution learning-based models, for example diffusion models, to solve this challenge as these methods are designed to fit the distribution of training data as close as possible. In this paper, we show that Bayesian flow network is capable of effortlessly generating high quality out-of-distribution samples that meet several scenarios. We introduce a semi-autoregressive training/sampling method that helps to enhance the model performance and surpass the state-of-the-art models.
Phase diagram and eigenvalue dynamics of stochastic gradient descent in multilayer neural networks
Hyperparameter tuning is one of the essential steps to guarantee the convergence of machine learning models. We argue that intuition about the optimal choice of hyperparameters for stochastic gradient descent can be obtained by studying a neural network's phase diagram, in which each phase is characterised by distinctive dynamics of the singular values of weight matrices. Taking inspiration from disordered systems, we start from the observation that the loss landscape of a multilayer neural network with mean squared error can be interpreted as a disordered system in feature space, where the learnt features are mapped to soft spin degrees of freedom, the initial variance of the weight matrices is interpreted as the strength of the disorder, and temperature is given by the ratio of the learning rate and the batch size. As the model is trained, three phases can be identified, in which the dynamics of weight matrices is qualitatively different. Employing a Langevin equation for stochastic gradient descent, previously derived using Dyson Brownian motion, we demonstrate that the three dynamical regimes can be classified effectively, providing practical guidance for the choice of hyperparameters of the optimiser.
Otter-Knowledge: benchmarks of multimodal knowledge graph representation learning from different sources for drug discovery
Recent research in representation learning utilizes large databases of proteins or molecules to acquire knowledge of drug and protein structures through unsupervised learning techniques. These pre-trained representations have proven to significantly enhance the accuracy of subsequent tasks, such as predicting the affinity between drugs and target proteins. In this study, we demonstrate that by incorporating knowledge graphs from diverse sources and modalities into the sequences or SMILES representation, we can further enrich the representation and achieve state-of-the-art results on established benchmark datasets. We provide preprocessed and integrated data obtained from 7 public sources, which encompass over 30M triples. Additionally, we make available the pre-trained models based on this data, along with the reported outcomes of their performance on three widely-used benchmark datasets for drug-target binding affinity prediction found in the Therapeutic Data Commons (TDC) benchmarks. Additionally, we make the source code for training models on benchmark datasets publicly available. Our objective in releasing these pre-trained models, accompanied by clean data for model pretraining and benchmark results, is to encourage research in knowledge-enhanced representation learning.



Data-Juicer Sandbox: A Comprehensive Suite for Multimodal Data-Model Co-development
The emergence of large-scale multi-modal generative models has drastically advanced artificial intelligence, introducing unprecedented levels of performance and functionality. However, optimizing these models remains challenging due to historically isolated paths of model-centric and data-centric developments, leading to suboptimal outcomes and inefficient resource utilization. In response, we present a novel sandbox suite tailored for integrated data-model co-development. This sandbox provides a comprehensive experimental platform, enabling rapid iteration and insight-driven refinement of both data and models. Our proposed "Probe-Analyze-Refine" workflow, validated through applications on state-of-the-art LLaVA-like and DiT based models, yields significant performance boosts, such as topping the VBench leaderboard. We also uncover fruitful insights gleaned from exhaustive benchmarks, shedding light on the critical interplay between data quality, diversity, and model behavior. With the hope of fostering deeper understanding and future progress in multi-modal data and generative modeling, our codes, datasets, and models are maintained and accessible at https://github.com/modelscope/data-juicer/blob/main/docs/Sandbox.md.

Transport meets Variational Inference: Controlled Monte Carlo Diffusions
Connecting optimal transport and variational inference, we present a principled and systematic framework for sampling and generative modelling centred around divergences on path space. Our work culminates in the development of the Controlled Monte Carlo Diffusion sampler (CMCD) for Bayesian computation, a score-based annealing technique that crucially adapts both forward and backward dynamics in a diffusion model. On the way, we clarify the relationship between the EM-algorithm and iterative proportional fitting (IPF) for Schr{\"o}dinger bridges, deriving as well a regularised objective that bypasses the iterative bottleneck of standard IPF-updates. Finally, we show that CMCD has a strong foundation in the Jarzinsky and Crooks identities from statistical physics, and that it convincingly outperforms competing approaches across a wide array of experiments.
The information-theoretic foundation of thermodynamic work extraction
In this paper I apply newly-proposed information-theoretic principles to thermodynamic work extraction. I show that if it is possible to extract work deterministically from a physical system prepared in any one of a set of states, then those states must be distinguishable from one another. This result is formulated independently of scale and of particular dynamical laws; it also provides a novel connection between thermodynamics and information theory, established via the law of conservation of energy (rather than the second law of thermodynamics). Albeit compatible with these conclusions, existing thermodynamics approaches cannot provide a result of such generality, because they are scale-dependent (relying on ensembles or coarse-graining) or tied to particular dynamical laws. This paper thus provides a broader foundation for thermodynamics, with implications for the theory of von Neumann's universal constructor
Optimistic Games for Combinatorial Bayesian Optimization with Application to Protein Design
Bayesian optimization (BO) is a powerful framework to optimize black-box expensive-to-evaluate functions via sequential interactions. In several important problems (e.g. drug discovery, circuit design, neural architecture search, etc.), though, such functions are defined over large combinatorial and unstructured spaces. This makes existing BO algorithms not feasible due to the intractable maximization of the acquisition function over these domains. To address this issue, we propose GameOpt, a novel game-theoretical approach to combinatorial BO. GameOpt establishes a cooperative game between the different optimization variables, and selects points that are game equilibria of an upper confidence bound acquisition function. These are stable configurations from which no variable has an incentive to deviate- analog to local optima in continuous domains. Crucially, this allows us to efficiently break down the complexity of the combinatorial domain into individual decision sets, making GameOpt scalable to large combinatorial spaces. We demonstrate the application of GameOpt to the challenging protein design problem and validate its performance on four real-world protein datasets. Each protein can take up to 20^{X} possible configurations, where X is the length of a protein, making standard BO methods infeasible. Instead, our approach iteratively selects informative protein configurations and very quickly discovers highly active protein variants compared to other baselines.

Representation Learning with Multi-Step Inverse Kinematics: An Efficient and Optimal Approach to Rich-Observation RL
We study the design of sample-efficient algorithms for reinforcement learning in the presence of rich, high-dimensional observations, formalized via the Block MDP problem. Existing algorithms suffer from either 1) computational intractability, 2) strong statistical assumptions that are not necessarily satisfied in practice, or 3) suboptimal sample complexity. We address these issues by providing the first computationally efficient algorithm that attains rate-optimal sample complexity with respect to the desired accuracy level, with minimal statistical assumptions. Our algorithm, MusIK, combines systematic exploration with representation learning based on multi-step inverse kinematics, a learning objective in which the aim is to predict the learner's own action from the current observation and observations in the (potentially distant) future. MusIK is simple and flexible, and can efficiently take advantage of general-purpose function approximation. Our analysis leverages several new techniques tailored to non-optimistic exploration algorithms, which we anticipate will find broader use.
Adaptive Pruning for Increased Robustness and Reduced Computational Overhead in Gaussian Process Accelerated Saddle Point Searches
Gaussian process (GP) regression provides a strategy for accelerating saddle point searches on high-dimensional energy surfaces by reducing the number of times the energy and its derivatives with respect to atomic coordinates need to be evaluated. The computational overhead in the hyperparameter optimization can, however, be large and make the approach inefficient. Failures can also occur if the search ventures too far into regions that are not represented well enough by the GP model. Here, these challenges are resolved by using geometry-aware optimal transport measures and an active pruning strategy using a summation over Wasserstein-1 distances for each atom-type in farthest-point sampling, selecting a fixed-size subset of geometrically diverse configurations to avoid rapidly increasing cost of GP updates as more observations are made. Stability is enhanced by permutation-invariant metric that provides a reliable trust radius for early-stopping and a logarithmic barrier penalty for the growth of the signal variance. These physically motivated algorithmic changes prove their efficacy by reducing to less than a half the mean computational time on a set of 238 challenging configurations from a previously published data set of chemical reactions. With these improvements, the GP approach is established as, a robust and scalable algorithm for accelerating saddle point searches when the evaluation of the energy and atomic forces requires significant computational effort.
ID and OOD Performance Are Sometimes Inversely Correlated on Real-world Datasets
Several studies have compared the in-distribution (ID) and out-of-distribution (OOD) performance of models in computer vision and NLP. They report a frequent positive correlation and some surprisingly never even observe an inverse correlation indicative of a necessary trade-off. The possibility of inverse patterns is important to determine whether ID performance can serve as a proxy for OOD generalization capabilities. This paper shows with multiple datasets that inverse correlations between ID and OOD performance do happen in real-world data - not only in theoretical worst-case settings. We also explain theoretically how these cases can arise even in a minimal linear setting, and why past studies could miss such cases due to a biased selection of models. Our observations lead to recommendations that contradict those found in much of the current literature. - High OOD performance sometimes requires trading off ID performance. - Focusing on ID performance alone may not lead to optimal OOD performance. It may produce diminishing (eventually negative) returns in OOD performance. - In these cases, studies on OOD generalization that use ID performance for model selection (a common recommended practice) will necessarily miss the best-performing models, making these studies blind to a whole range of phenomena.
Quantum Monte Carlo simulations in the restricted Hilbert space of Rydberg atom arrays
Rydberg atom arrays have emerged as a powerful platform to simulate a number of exotic quantum ground states and phase transitions. To verify these capabilities numerically, we develop a versatile quantum Monte Carlo sampling technique which operates in the reduced Hilbert space generated by enforcing the constraint of a Rydberg blockade. We use the framework of stochastic series expansion and show that in the restricted space, the configuration space of operator strings can be understood as a hard rod gas in d+1 dimensions. We use this mapping to develop cluster algorithms which can be visualized as various non-local movements of rods. We study the efficiency of each of our updates individually and collectively. To elucidate the utility of the algorithm, we show that it can efficiently generate the phase diagram of a Rydberg atom array, to temperatures much smaller than all energy scales involved, on a Kagom\'e link lattice. This is of broad interest as the presence of a Z_2 spin liquid has been hypothesized recently.
On the Existence of Universal Lottery Tickets
The lottery ticket hypothesis conjectures the existence of sparse subnetworks of large randomly initialized deep neural networks that can be successfully trained in isolation. Recent work has experimentally observed that some of these tickets can be practically reused across a variety of tasks, hinting at some form of universality. We formalize this concept and theoretically prove that not only do such universal tickets exist but they also do not require further training. Our proofs introduce a couple of technical innovations related to pruning for strong lottery tickets, including extensions of subset sum results and a strategy to leverage higher amounts of depth. Our explicit sparse constructions of universal function families might be of independent interest, as they highlight representational benefits induced by univariate convolutional architectures.
Str2Str: A Score-based Framework for Zero-shot Protein Conformation Sampling
The dynamic nature of proteins is crucial for determining their biological functions and properties, for which Monte Carlo (MC) and molecular dynamics (MD) simulations stand as predominant tools to study such phenomena. By utilizing empirically derived force fields, MC or MD simulations explore the conformational space through numerically evolving the system via Markov chain or Newtonian mechanics. However, the high-energy barrier of the force fields can hamper the exploration of both methods by the rare event, resulting in inadequately sampled ensemble without exhaustive running. Existing learning-based approaches perform direct sampling yet heavily rely on target-specific simulation data for training, which suffers from high data acquisition cost and poor generalizability. Inspired by simulated annealing, we propose Str2Str, a novel structure-to-structure translation framework capable of zero-shot conformation sampling with roto-translation equivariant property. Our method leverages an amortized denoising score matching objective trained on general crystal structures and has no reliance on simulation data during both training and inference. Experimental results across several benchmarking protein systems demonstrate that Str2Str outperforms previous state-of-the-art generative structure prediction models and can be orders of magnitude faster compared to long MD simulations. Our open-source implementation is available at https://github.com/lujiarui/Str2Str


Oracle Efficient Algorithms for Groupwise Regret
We study the problem of online prediction, in which at each time step t, an individual x_t arrives, whose label we must predict. Each individual is associated with various groups, defined based on their features such as age, sex, race etc., which may intersect. Our goal is to make predictions that have regret guarantees not just overall but also simultaneously on each sub-sequence comprised of the members of any single group. Previous work such as [Blum & Lykouris] and [Lee et al] provide attractive regret guarantees for these problems; however, these are computationally intractable on large model classes. We show that a simple modification of the sleeping experts technique of [Blum & Lykouris] yields an efficient reduction to the well-understood problem of obtaining diminishing external regret absent group considerations. Our approach gives similar regret guarantees compared to [Blum & Lykouris]; however, we run in time linear in the number of groups, and are oracle-efficient in the hypothesis class. This in particular implies that our algorithm is efficient whenever the number of groups is polynomially bounded and the external-regret problem can be solved efficiently, an improvement on [Blum & Lykouris]'s stronger condition that the model class must be small. Our approach can handle online linear regression and online combinatorial optimization problems like online shortest paths. Beyond providing theoretical regret bounds, we evaluate this algorithm with an extensive set of experiments on synthetic data and on two real data sets -- Medical costs and the Adult income dataset, both instantiated with intersecting groups defined in terms of race, sex, and other demographic characteristics. We find that uniformly across groups, our algorithm gives substantial error improvements compared to running a standard online linear regression algorithm with no groupwise regret guarantees.
Knowledge-informed Molecular Learning: A Survey on Paradigm Transfer
Machine learning, notably deep learning, has significantly propelled molecular investigations within the biochemical sphere. Traditionally, modeling for such research has centered around a handful of paradigms. For instance, the prediction paradigm is frequently deployed for tasks such as molecular property prediction. To enhance the generation and decipherability of purely data-driven models, scholars have integrated biochemical domain knowledge into these molecular study models. This integration has sparked a surge in paradigm transfer, which is solving one molecular learning task by reformulating it as another one. With the emergence of Large Language Models, these paradigms have demonstrated an escalating trend towards harmonized unification. In this work, we delineate a literature survey focused on knowledge-informed molecular learning from the perspective of paradigm transfer. We classify the paradigms, scrutinize their methodologies, and dissect the contribution of domain knowledge. Moreover, we encapsulate prevailing trends and identify intriguing avenues for future exploration in molecular learning.



Dropout-Based Rashomon Set Exploration for Efficient Predictive Multiplicity Estimation
Predictive multiplicity refers to the phenomenon in which classification tasks may admit multiple competing models that achieve almost-equally-optimal performance, yet generate conflicting outputs for individual samples. This presents significant concerns, as it can potentially result in systemic exclusion, inexplicable discrimination, and unfairness in practical applications. Measuring and mitigating predictive multiplicity, however, is computationally challenging due to the need to explore all such almost-equally-optimal models, known as the Rashomon set, in potentially huge hypothesis spaces. To address this challenge, we propose a novel framework that utilizes dropout techniques for exploring models in the Rashomon set. We provide rigorous theoretical derivations to connect the dropout parameters to properties of the Rashomon set, and empirically evaluate our framework through extensive experimentation. Numerical results show that our technique consistently outperforms baselines in terms of the effectiveness of predictive multiplicity metric estimation, with runtime speedup up to 20times sim 5000times. With efficient Rashomon set exploration and metric estimation, mitigation of predictive multiplicity is then achieved through dropout ensemble and model selection.

Run-Off Election: Improved Provable Defense against Data Poisoning Attacks
In data poisoning attacks, an adversary tries to change a model's prediction by adding, modifying, or removing samples in the training data. Recently, ensemble-based approaches for obtaining provable defenses against data poisoning have been proposed where predictions are done by taking a majority vote across multiple base models. In this work, we show that merely considering the majority vote in ensemble defenses is wasteful as it does not effectively utilize available information in the logits layers of the base models. Instead, we propose Run-Off Election (ROE), a novel aggregation method based on a two-round election across the base models: In the first round, models vote for their preferred class and then a second, Run-Off election is held between the top two classes in the first round. Based on this approach, we propose DPA+ROE and FA+ROE defense methods based on Deep Partition Aggregation (DPA) and Finite Aggregation (FA) approaches from prior work. We evaluate our methods on MNIST, CIFAR-10, and GTSRB and obtain improvements in certified accuracy by up to 3%-4%. Also, by applying ROE on a boosted version of DPA, we gain improvements around 12%-27% comparing to the current state-of-the-art, establishing a new state-of-the-art in (pointwise) certified robustness against data poisoning. In many cases, our approach outperforms the state-of-the-art, even when using 32 times less computational power.


Differentiable Simulations for Enhanced Sampling of Rare Events
Simulating rare events, such as the transformation of a reactant into a product in a chemical reaction typically requires enhanced sampling techniques that rely on heuristically chosen collective variables (CVs). We propose using differentiable simulations (DiffSim) for the discovery and enhanced sampling of chemical transformations without a need to resort to preselected CVs, using only a distance metric. Reaction path discovery and estimation of the biasing potential that enhances the sampling are merged into a single end-to-end problem that is solved by path-integral optimization. This is achieved by introducing multiple improvements over standard DiffSim such as partial backpropagation and graph mini-batching making DiffSim training stable and efficient. The potential of DiffSim is demonstrated in the successful discovery of transition paths for the Muller-Brown model potential as well as a benchmark chemical system - alanine dipeptide.
Independent-Set Design of Experiments for Estimating Treatment and Spillover Effects under Network Interference
Interference is ubiquitous when conducting causal experiments over networks. Except for certain network structures, causal inference on the network in the presence of interference is difficult due to the entanglement between the treatment assignments and the interference levels. In this article, we conduct causal inference under interference on an observed, sparse but connected network, and we propose a novel design of experiments based on an independent set. Compared to conventional designs, the independent-set design focuses on an independent subset of data and controls their interference exposures through the assignments to the rest (auxiliary set). We provide a lower bound on the size of the independent set from a greedy algorithm , and justify the theoretical performance of estimators under the proposed design. Our approach is capable of estimating both spillover effects and treatment effects. We justify its superiority over conventional methods and illustrate the empirical performance through simulations.
Theoretical Guarantees of Learning Ensembling Strategies with Applications to Time Series Forecasting
Ensembling is among the most popular tools in machine learning (ML) due to its effectiveness in minimizing variance and thus improving generalization. Most ensembling methods for black-box base learners fall under the umbrella of "stacked generalization," namely training an ML algorithm that takes the inferences from the base learners as input. While stacking has been widely applied in practice, its theoretical properties are poorly understood. In this paper, we prove a novel result, showing that choosing the best stacked generalization from a (finite or finite-dimensional) family of stacked generalizations based on cross-validated performance does not perform "much worse" than the oracle best. Our result strengthens and significantly extends the results in Van der Laan et al. (2007). Inspired by the theoretical analysis, we further propose a particular family of stacked generalizations in the context of probabilistic forecasting, each one with a different sensitivity for how much the ensemble weights are allowed to vary across items, timestamps in the forecast horizon, and quantiles. Experimental results demonstrate the performance gain of the proposed method.

Revisiting Link Prediction: A Data Perspective
Link prediction, a fundamental task on graphs, has proven indispensable in various applications, e.g., friend recommendation, protein analysis, and drug interaction prediction. However, since datasets span a multitude of domains, they could have distinct underlying mechanisms of link formation. Evidence in existing literature underscores the absence of a universally best algorithm suitable for all datasets. In this paper, we endeavor to explore principles of link prediction across diverse datasets from a data-centric perspective. We recognize three fundamental factors critical to link prediction: local structural proximity, global structural proximity, and feature proximity. We then unearth relationships among those factors where (i) global structural proximity only shows effectiveness when local structural proximity is deficient. (ii) The incompatibility can be found between feature and structural proximity. Such incompatibility leads to GNNs for Link Prediction (GNN4LP) consistently underperforming on edges where the feature proximity factor dominates. Inspired by these new insights from a data perspective, we offer practical instruction for GNN4LP model design and guidelines for selecting appropriate benchmark datasets for more comprehensive evaluations.
Timewarp: Transferable Acceleration of Molecular Dynamics by Learning Time-Coarsened Dynamics
Molecular dynamics (MD) simulation is a widely used technique to simulate molecular systems, most commonly at the all-atom resolution where equations of motion are integrated with timesteps on the order of femtoseconds (1fs=10^{-15}s). MD is often used to compute equilibrium properties, which requires sampling from an equilibrium distribution such as the Boltzmann distribution. However, many important processes, such as binding and folding, occur over timescales of milliseconds or beyond, and cannot be efficiently sampled with conventional MD. Furthermore, new MD simulations need to be performed for each molecular system studied. We present Timewarp, an enhanced sampling method which uses a normalising flow as a proposal distribution in a Markov chain Monte Carlo method targeting the Boltzmann distribution. The flow is trained offline on MD trajectories and learns to make large steps in time, simulating the molecular dynamics of 10^{5} - 10^{6}:fs. Crucially, Timewarp is transferable between molecular systems: once trained, we show that it generalises to unseen small peptides (2-4 amino acids) at all-atom resolution, exploring their metastable states and providing wall-clock acceleration of sampling compared to standard MD. Our method constitutes an important step towards general, transferable algorithms for accelerating MD.
On Invariance Penalties for Risk Minimization
The Invariant Risk Minimization (IRM) principle was first proposed by Arjovsky et al. [2019] to address the domain generalization problem by leveraging data heterogeneity from differing experimental conditions. Specifically, IRM seeks to find a data representation under which an optimal classifier remains invariant across all domains. Despite the conceptual appeal of IRM, the effectiveness of the originally proposed invariance penalty has recently been brought into question. In particular, there exists counterexamples for which that invariance penalty can be arbitrarily small for non-invariant data representations. We propose an alternative invariance penalty by revisiting the Gramian matrix of the data representation. We discuss the role of its eigenvalues in the relationship between the risk and the invariance penalty, and demonstrate that it is ill-conditioned for said counterexamples. The proposed approach is guaranteed to recover an invariant representation for linear settings under mild non-degeneracy conditions. Its effectiveness is substantiated by experiments on DomainBed and InvarianceUnitTest, two extensive test beds for domain generalization.
Von Mises Mixture Distributions for Molecular Conformation Generation
Molecules are frequently represented as graphs, but the underlying 3D molecular geometry (the locations of the atoms) ultimately determines most molecular properties. However, most molecules are not static and at room temperature adopt a wide variety of geometries or conformations. The resulting distribution on geometries p(x) is known as the Boltzmann distribution, and many molecular properties are expectations computed under this distribution. Generating accurate samples from the Boltzmann distribution is therefore essential for computing these expectations accurately. Traditional sampling-based methods are computationally expensive, and most recent machine learning-based methods have focused on identifying modes in this distribution rather than generating true samples. Generating such samples requires capturing conformational variability, and it has been widely recognized that the majority of conformational variability in molecules arises from rotatable bonds. In this work, we present VonMisesNet, a new graph neural network that captures conformational variability via a variational approximation of rotatable bond torsion angles as a mixture of von Mises distributions. We demonstrate that VonMisesNet can generate conformations for arbitrary molecules in a way that is both physically accurate with respect to the Boltzmann distribution and orders of magnitude faster than existing sampling methods.
Deep Confident Steps to New Pockets: Strategies for Docking Generalization
Accurate blind docking has the potential to lead to new biological breakthroughs, but for this promise to be realized, docking methods must generalize well across the proteome. Existing benchmarks, however, fail to rigorously assess generalizability. Therefore, we develop DockGen, a new benchmark based on the ligand-binding domains of proteins, and we show that existing machine learning-based docking models have very weak generalization abilities. We carefully analyze the scaling laws of ML-based docking and show that, by scaling data and model size, as well as integrating synthetic data strategies, we are able to significantly increase the generalization capacity and set new state-of-the-art performance across benchmarks. Further, we propose Confidence Bootstrapping, a new training paradigm that solely relies on the interaction between diffusion and confidence models and exploits the multi-resolution generation process of diffusion models. We demonstrate that Confidence Bootstrapping significantly improves the ability of ML-based docking methods to dock to unseen protein classes, edging closer to accurate and generalizable blind docking methods.
Composition and Control with Distilled Energy Diffusion Models and Sequential Monte Carlo
Diffusion models may be formulated as a time-indexed sequence of energy-based models, where the score corresponds to the negative gradient of an energy function. As opposed to learning the score directly, an energy parameterization is attractive as the energy itself can be used to control generation via Monte Carlo samplers. Architectural constraints and training instability in energy parameterized models have so far yielded inferior performance compared to directly approximating the score or denoiser. We address these deficiencies by introducing a novel training regime for the energy function through distillation of pre-trained diffusion models, resembling a Helmholtz decomposition of the score vector field. We further showcase the synergies between energy and score by casting the diffusion sampling procedure as a Feynman Kac model where sampling is controlled using potentials from the learnt energy functions. The Feynman Kac model formalism enables composition and low temperature sampling through sequential Monte Carlo.
A Learnable Prior Improves Inverse Tumor Growth Modeling
Biophysical modeling, particularly involving partial differential equations (PDEs), offers significant potential for tailoring disease treatment protocols to individual patients. However, the inverse problem-solving aspect of these models presents a substantial challenge, either due to the high computational requirements of model-based approaches or the limited robustness of deep learning (DL) methods. We propose a novel framework that leverages the unique strengths of both approaches in a synergistic manner. Our method incorporates a DL ensemble for initial parameter estimation, facilitating efficient downstream evolutionary sampling initialized with this DL-based prior. We showcase the effectiveness of integrating a rapid deep-learning algorithm with a high-precision evolution strategy in estimating brain tumor cell concentrations from magnetic resonance images. The DL-Prior plays a pivotal role, significantly constraining the effective sampling-parameter space. This reduction results in a fivefold convergence acceleration and a Dice-score of 95%
Learning Smooth and Expressive Interatomic Potentials for Physical Property Prediction
Machine learning interatomic potentials (MLIPs) have become increasingly effective at approximating quantum mechanical calculations at a fraction of the computational cost. However, lower errors on held out test sets do not always translate to improved results on downstream physical property prediction tasks. In this paper, we propose testing MLIPs on their practical ability to conserve energy during molecular dynamic simulations. If passed, improved correlations are found between test errors and their performance on physical property prediction tasks. We identify choices which may lead to models failing this test, and use these observations to improve upon highly-expressive models. The resulting model, eSEN, provides state-of-the-art results on a range of physical property prediction tasks, including materials stability prediction, thermal conductivity prediction, and phonon calculations.


Impact Assessment of Missing Data in Model Predictions for Earth Observation Applications
Earth observation (EO) applications involving complex and heterogeneous data sources are commonly approached with machine learning models. However, there is a common assumption that data sources will be persistently available. Different situations could affect the availability of EO sources, like noise, clouds, or satellite mission failures. In this work, we assess the impact of missing temporal and static EO sources in trained models across four datasets with classification and regression tasks. We compare the predictive quality of different methods and find that some are naturally more robust to missing data. The Ensemble strategy, in particular, achieves a prediction robustness up to 100%. We evidence that missing scenarios are significantly more challenging in regression than classification tasks. Finally, we find that the optical view is the most critical view when it is missing individually.

Solving Inverse Problems via Diffusion-Based Priors: An Approximation-Free Ensemble Sampling Approach
Diffusion models (DMs) have proven to be effective in modeling high-dimensional distributions, leading to their widespread adoption for representing complex priors in Bayesian inverse problems (BIPs). However, current DM-based posterior sampling methods proposed for solving common BIPs rely on heuristic approximations to the generative process. To exploit the generative capability of DMs and avoid the usage of such approximations, we propose an ensemble-based algorithm that performs posterior sampling without the use of heuristic approximations. Our algorithm is motivated by existing works that combine DM-based methods with the sequential Monte Carlo (SMC) method. By examining how the prior evolves through the diffusion process encoded by the pre-trained score function, we derive a modified partial differential equation (PDE) governing the evolution of the corresponding posterior distribution. This PDE includes a modified diffusion term and a reweighting term, which can be simulated via stochastic weighted particle methods. Theoretically, we prove that the error between the true posterior distribution can be bounded in terms of the training error of the pre-trained score function and the number of particles in the ensemble. Empirically, we validate our algorithm on several inverse problems in imaging to show that our method gives more accurate reconstructions compared to existing DM-based methods.
DISCO: Diversifying Sample Condensation for Efficient Model Evaluation
Evaluating modern machine learning models has become prohibitively expensive. Benchmarks such as LMMs-Eval and HELM demand thousands of GPU hours per model. Costly evaluation reduces inclusivity, slows the cycle of innovation, and worsens environmental impact. The typical approach follows two steps. First, select an anchor subset of data. Second, train a mapping from the accuracy on this subset to the final test result. The drawback is that anchor selection depends on clustering, which can be complex and sensitive to design choices. We argue that promoting diversity among samples is not essential; what matters is to select samples that maximise diversity in model responses. Our method, Diversifying Sample Condensation (DISCO), selects the top-k samples with the greatest model disagreements. This uses greedy, sample-wise statistics rather than global clustering. The approach is conceptually simpler. From a theoretical view, inter-model disagreement provides an information-theoretically optimal rule for such greedy selection. DISCO shows empirical gains over prior methods, achieving state-of-the-art results in performance prediction across MMLU, Hellaswag, Winogrande, and ARC. Code is available here: https://github.com/arubique/disco-public.

MDNS: Masked Diffusion Neural Sampler via Stochastic Optimal Control
We study the problem of learning a neural sampler to generate samples from discrete state spaces where the target probability mass function piproptoe^{-U} is known up to a normalizing constant, which is an important task in fields such as statistical physics, machine learning, combinatorial optimization, etc. To better address this challenging task when the state space has a large cardinality and the distribution is multi-modal, we propose Masked Diffusion Neural Sampler (MDNS), a novel framework for training discrete neural samplers by aligning two path measures through a family of learning objectives, theoretically grounded in the stochastic optimal control of the continuous-time Markov chains. We validate the efficiency and scalability of MDNS through extensive experiments on various distributions with distinct statistical properties, where MDNS learns to accurately sample from the target distributions despite the extremely high problem dimensions and outperforms other learning-based baselines by a large margin. A comprehensive study of ablations and extensions is also provided to demonstrate the efficacy and potential of the proposed framework.
Effective Diversity in Population Based Reinforcement Learning
Exploration is a key problem in reinforcement learning, since agents can only learn from data they acquire in the environment. With that in mind, maintaining a population of agents is an attractive method, as it allows data be collected with a diverse set of behaviors. This behavioral diversity is often boosted via multi-objective loss functions. However, those approaches typically leverage mean field updates based on pairwise distances, which makes them susceptible to cycling behaviors and increased redundancy. In addition, explicitly boosting diversity often has a detrimental impact on optimizing already fruitful behaviors for rewards. As such, the reward-diversity trade off typically relies on heuristics. Finally, such methods require behavioral representations, often handcrafted and domain specific. In this paper, we introduce an approach to optimize all members of a population simultaneously. Rather than using pairwise distance, we measure the volume of the entire population in a behavioral manifold, defined by task-agnostic behavioral embeddings. In addition, our algorithm Diversity via Determinants (DvD), adapts the degree of diversity during training using online learning techniques. We introduce both evolutionary and gradient-based instantiations of DvD and show they effectively improve exploration without reducing performance when better exploration is not required.
Predictable Compression Failures: Why Language Models Actually Hallucinate
Large language models perform near-Bayesian inference yet violate permutation invariance on exchangeable data. We resolve this by showing transformers minimize expected conditional description length (cross-entropy) over orderings, E_pi[ell(Y mid Gamma_pi(X))], which admits a Kolmogorov-complexity interpretation up to additive constants, rather than the permutation-invariant description length ell(Y mid X). This makes them Bayesian in expectation, not in realization. We derive (i) a Quantified Martingale Violation bound showing order-induced deviations scale as O(log n) with constants; (ii) the Expectation-level Decompression Law linking information budgets to reliability for Bernoulli predicates; and (iii) deployable planners (B2T/RoH/ISR) for answer/abstain decisions. Empirically, permutation dispersion follows a+bln n (Qwen2-7B b approx 0.377, Llama-3.1-8B b approx 0.147); permutation mixtures improve ground-truth likelihood/accuracy; and randomized dose-response shows hallucinations drop by sim 0.13 per additional nat. A pre-specified audit with a fixed ISR=1.0 achieves near-0\% hallucinations via calibrated refusal at 24\% abstention. The framework turns hallucinations into predictable compression failures and enables principled information budgeting.
The Price of Freedom: Exploring Expressivity and Runtime Tradeoffs in Equivariant Tensor Products
E(3)-equivariant neural networks have demonstrated success across a wide range of 3D modelling tasks. A fundamental operation in these networks is the tensor product, which interacts two geometric features in an equivariant manner to create new features. Due to the high computational complexity of the tensor product, significant effort has been invested to optimize the runtime of this operation. For example, Luo et al. (2024) recently proposed the Gaunt tensor product (GTP) which promises a significant speedup. In this work, we provide a careful, systematic analysis of a number of tensor product operations. In particular, we emphasize that different tensor products are not performing the same operation. The reported speedups typically come at the cost of expressivity. We introduce measures of expressivity and interactability to characterize these differences. In addition, we realized the original implementation of GTP can be greatly simplified by directly using a spherical grid at no cost in asymptotic runtime. This spherical grid approach is faster on our benchmarks and in actual training of the MACE interatomic potential by 30%. Finally, we provide the first systematic microbenchmarks of the various tensor product operations. We find that the theoretical runtime guarantees can differ wildly from empirical performance, demonstrating the need for careful application-specific benchmarking. Code is available at https://github.com/atomicarchitects/PriceofFreedom.
Adaptive Data-Knowledge Alignment in Genetic Perturbation Prediction
The transcriptional response to genetic perturbation reveals fundamental insights into complex cellular systems. While current approaches have made progress in predicting genetic perturbation responses, they provide limited biological understanding and cannot systematically refine existing knowledge. Overcoming these limitations requires an end-to-end integration of data-driven learning and existing knowledge. However, this integration is challenging due to inconsistencies between data and knowledge bases, such as noise, misannotation, and incompleteness. To address this challenge, we propose ALIGNED (Adaptive aLignment for Inconsistent Genetic kNowledgE and Data), a neuro-symbolic framework based on the Abductive Learning (ABL) paradigm. This end-to-end framework aligns neural and symbolic components and performs systematic knowledge refinement. We introduce a balanced consistency metric to evaluate the predictions' consistency against both data and knowledge. Our results show that ALIGNED outperforms state-of-the-art methods by achieving the highest balanced consistency, while also re-discovering biologically meaningful knowledge. Our work advances beyond existing methods to enable both the transparency and the evolution of mechanistic biological understanding.
Monge, Bregman and Occam: Interpretable Optimal Transport in High-Dimensions with Feature-Sparse Maps
Optimal transport (OT) theory focuses, among all maps T:R^drightarrow R^d that can morph a probability measure onto another, on those that are the ``thriftiest'', i.e. such that the averaged cost c(x, T(x)) between x and its image T(x) be as small as possible. Many computational approaches have been proposed to estimate such Monge maps when c is the ell_2^2 distance, e.g., using entropic maps [Pooladian'22], or neural networks [Makkuva'20, Korotin'20]. We propose a new model for transport maps, built on a family of translation invariant costs c(x, y):=h(x-y), where h:=1{2}|cdot|_2^2+tau and tau is a regularizer. We propose a generalization of the entropic map suitable for h, and highlight a surprising link tying it with the Bregman centroids of the divergence D_h generated by h, and the proximal operator of tau. We show that choosing a sparsity-inducing norm for tau results in maps that apply Occam's razor to transport, in the sense that the displacement vectors Delta(x):= T(x)-x they induce are sparse, with a sparsity pattern that varies depending on x. We showcase the ability of our method to estimate meaningful OT maps for high-dimensional single-cell transcription data, in the 34000-d space of gene counts for cells, without using dimensionality reduction, thus retaining the ability to interpret all displacements at the gene level.
Self-Guided Generation of Minority Samples Using Diffusion Models
We present a novel approach for generating minority samples that live on low-density regions of a data manifold. Our framework is built upon diffusion models, leveraging the principle of guided sampling that incorporates an arbitrary energy-based guidance during inference time. The key defining feature of our sampler lies in its self-contained nature, \ie, implementable solely with a pretrained model. This distinguishes our sampler from existing techniques that require expensive additional components (like external classifiers) for minority generation. Specifically, we first estimate the likelihood of features within an intermediate latent sample by evaluating a reconstruction loss w.r.t. its posterior mean. The generation then proceeds with the minimization of the estimated likelihood, thereby encouraging the emergence of minority features in the latent samples of subsequent timesteps. To further improve the performance of our sampler, we provide several time-scheduling techniques that properly manage the influence of guidance over inference steps. Experiments on benchmark real datasets demonstrate that our approach can greatly improve the capability of creating realistic low-likelihood minority instances over the existing techniques without the reliance on costly additional elements. Code is available at https://github.com/soobin-um/sg-minority.

Construction of simplicial complexes with prescribed degree-size sequences
We study the realizability of simplicial complexes with a given pair of integer sequences, representing the node degree distribution and the facet size distribution, respectively. While the s-uniform variant of the problem is NP-complete when s geq 3, we identify two populations of input sequences, most of which can be solved in polynomial time using a recursive algorithm that we contribute. Combining with a sampler for the simplicial configuration model [J.-G. Young et al., Phys. Rev. E 96, 032312 (2017)], we facilitate the efficient sampling of simplicial ensembles from arbitrary degree and size distributions. We find that, contrary to expectations based on dyadic networks, increasing the nodes' degrees reduces the number of loops in simplicial complexes. Our work unveils a fundamental constraint on the degree-size sequences and sheds light on further analysis of higher-order phenomena based on local structures.

Flow Matching for Discrete Systems: Efficient Free Energy Sampling Across Lattice Sizes and Temperatures
Generative models have advanced significantly in sampling material systems with continuous variables, such as atomistic structures. However, their application to discrete variables, like atom types or spin states, remains underexplored. In this work, we introduce a Boltzmann generator built on discrete flow matching, specifically tailored for systems with discrete phase-space coordinates (e.g., the Ising model or crystalline compounds). This approach enables a single model to sample free energy surfaces over a wide temperature range with minimal training overhead. In addition, the model generation is scalable to larger lattice sizes than those in the training set. We demonstrate the effectiveness of our approach on the 2D Ising model, showing efficient and reliable free energy sampling. This framework provides a scalable and computationally efficient solution for discrete coordinate systems and can be extended to sample the alchemical degrees of freedom in crystalline compounds.


Electron flow matching for generative reaction mechanism prediction obeying conservation laws
Central to our understanding of chemical reactivity is the principle of mass conservation, which is fundamental for ensuring physical consistency, balancing equations, and guiding reaction design. However, data-driven computational models for tasks such as reaction product prediction rarely abide by this most basic constraint. In this work, we recast the problem of reaction prediction as a problem of electron redistribution using the modern deep generative framework of flow matching. Our model, FlowER, overcomes limitations inherent in previous approaches by enforcing exact mass conservation, thereby resolving hallucinatory failure modes, recovering mechanistic reaction sequences for unseen substrate scaffolds, and generalizing effectively to out-of-domain reaction classes with extremely data-efficient fine-tuning. FlowER additionally enables estimation of thermodynamic or kinetic feasibility and manifests a degree of chemical intuition in reaction prediction tasks. This inherently interpretable framework represents a significant step in bridging the gap between predictive accuracy and mechanistic understanding in data-driven reaction outcome prediction.


A General Framework for Inference-time Scaling and Steering of Diffusion Models
Diffusion models produce impressive results in modalities ranging from images and video to protein design and text. However, generating samples with user-specified properties remains a challenge. Recent research proposes fine-tuning models to maximize rewards that capture desired properties, but these methods require expensive training and are prone to mode collapse. In this work, we propose Feynman Kac (FK) steering, an inference-time framework for steering diffusion models with reward functions. FK steering works by sampling a system of multiple interacting diffusion processes, called particles, and resampling particles at intermediate steps based on scores computed using functions called potentials. Potentials are defined using rewards for intermediate states and are selected such that a high value indicates that the particle will yield a high-reward sample. We explore various choices of potentials, intermediate rewards, and samplers. We evaluate FK steering on text-to-image and text diffusion models. For steering text-to-image models with a human preference reward, we find that FK steering a 0.8B parameter model outperforms a 2.6B parameter fine-tuned model on prompt fidelity, with faster sampling and no training. For steering text diffusion models with rewards for text quality and specific text attributes, we find that FK steering generates lower perplexity, more linguistically acceptable outputs and enables gradient-free control of attributes like toxicity. Our results demonstrate that inference-time scaling and steering of diffusion models, even with off-the-shelf rewards, can provide significant sample quality gains and controllability benefits. Code is available at https://github.com/zacharyhorvitz/Fk-Diffusion-Steering .

Learning to engineer protein flexibility
Generative machine learning models are increasingly being used to design novel proteins for therapeutic and biotechnological applications. However, the current methods mostly focus on the design of proteins with a fixed backbone structure, which leads to their limited ability to account for protein flexibility, one of the crucial properties for protein function. Learning to engineer protein flexibility is problematic because the available data are scarce, heterogeneous, and costly to obtain using computational as well as experimental methods. Our contributions to address this problem are three-fold. First, we comprehensively compare methods for quantifying protein flexibility and identify data relevant to learning. Second, we design and train flexibility predictors utilizing sequential or both sequential and structural information on the input. We overcome the data scarcity issue by leveraging a pre-trained protein language model. Third, we introduce a method for fine-tuning a protein inverse folding model to steer it toward desired flexibility in specified regions. We demonstrate that our method Flexpert-Design enables guidance of inverse folding models toward increased flexibility. This opens up new possibilities for protein flexibility engineering and the development of proteins with enhanced biological activities.